Signaling in action
TL;DR:
Our paper in eLife modeling G protein activation is out! Check it out here
A brief summary of our paper was also published as an eLife digest!
Introduction and Significance
To communicate and respond to our environment (i.e. via vision, smell, or taste), our cells rely on signaling cascades made up of groups of proteins that activate and deactivate one another, much like a circuit.
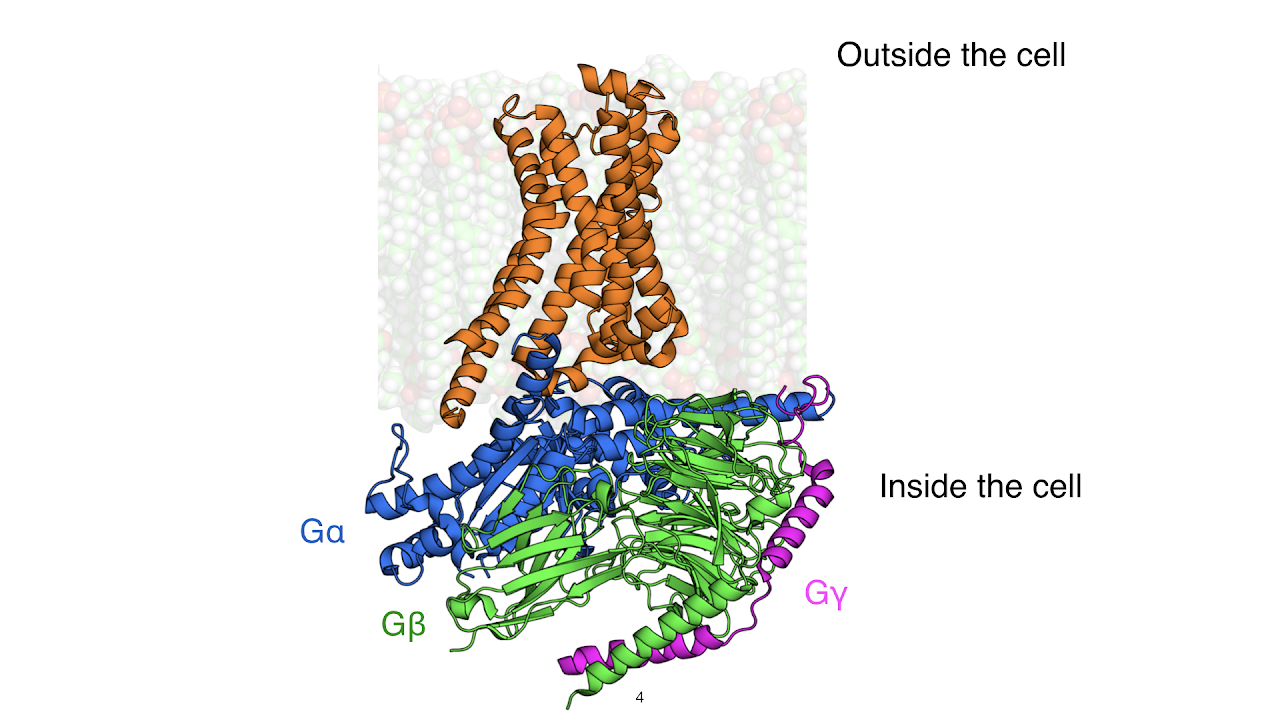
Many cascades in our body involve heterotrimeric G proteins, which have three components named G-alpha (Gα), G-beta (Gβ), and G-gamma (Gγ). Gα is a critical member of this trio, acting as a switch that turns on upon receiving a signal from a membrane protein receptor. This switch-like behavior of Gα is critical for life; both overactive and under-active Gα proteins have been linked to cancer, heart disease, and other disorders. However, no current therapies target Gα, despite the fact that roughly 40% of drugs on the market target other components of G-protein signaling cascades.
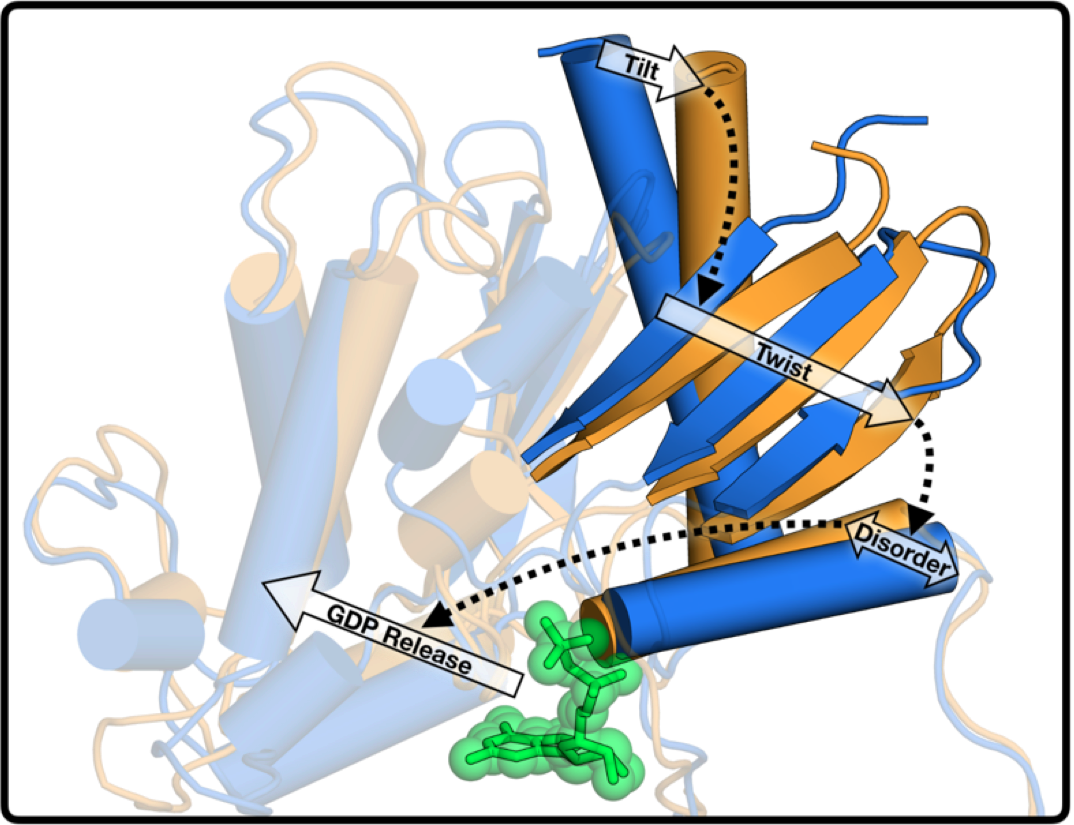
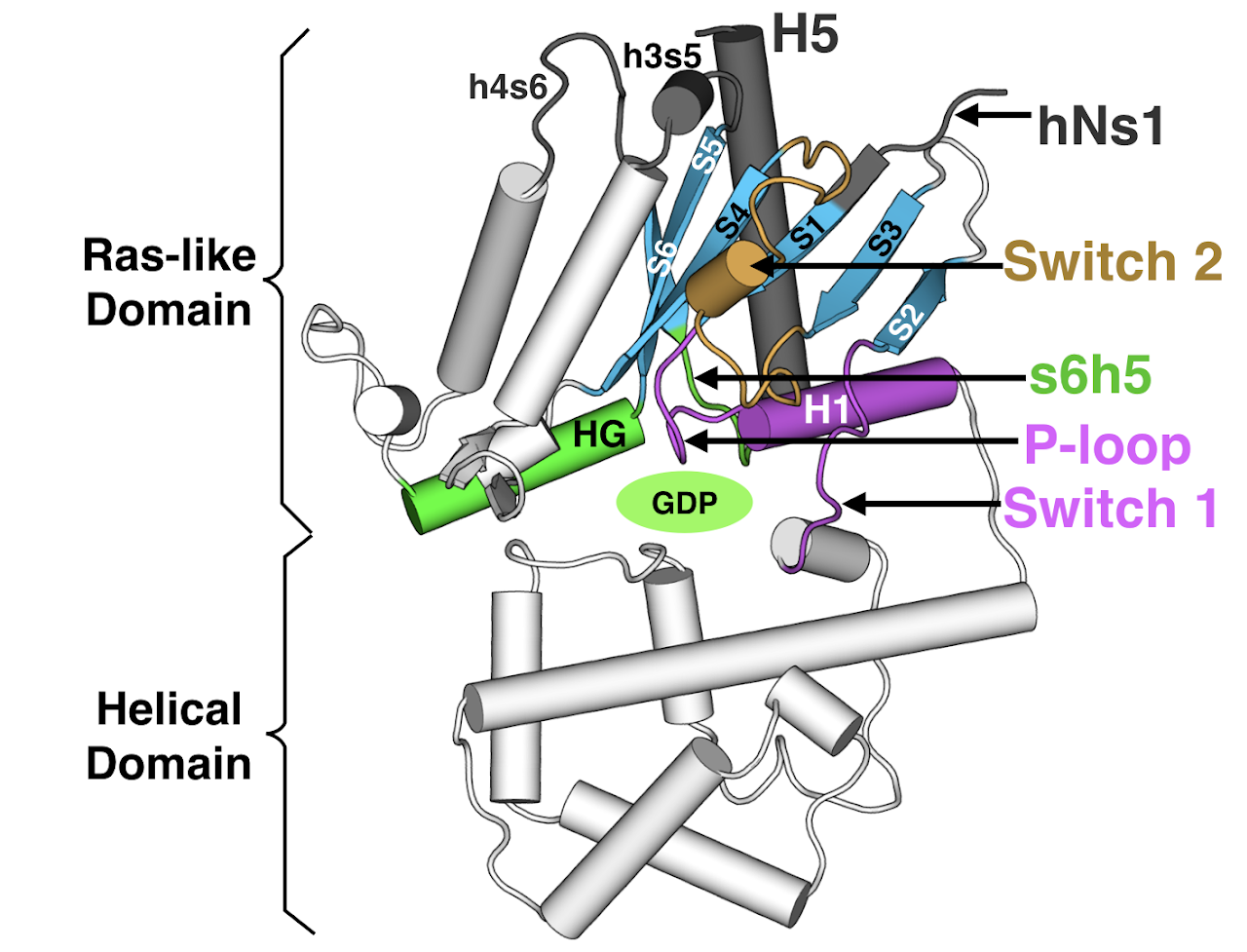
Understanding the structural changes governing the switch-like behavior of Gα would shed light into how signals are transmitted within our cells. When Gα is off, it is bound to the small molecule Guanosine diphosphate (GDP). To turn on, Gα must release GDP to bind Guanosine triphosphate (GTP). However, the details of GDP release from Gα, the critical step of Gα activation, remain unclear. This is the focus of our paper - what atomic motions and molecular changes drive GDP release during the process of Gα activation?
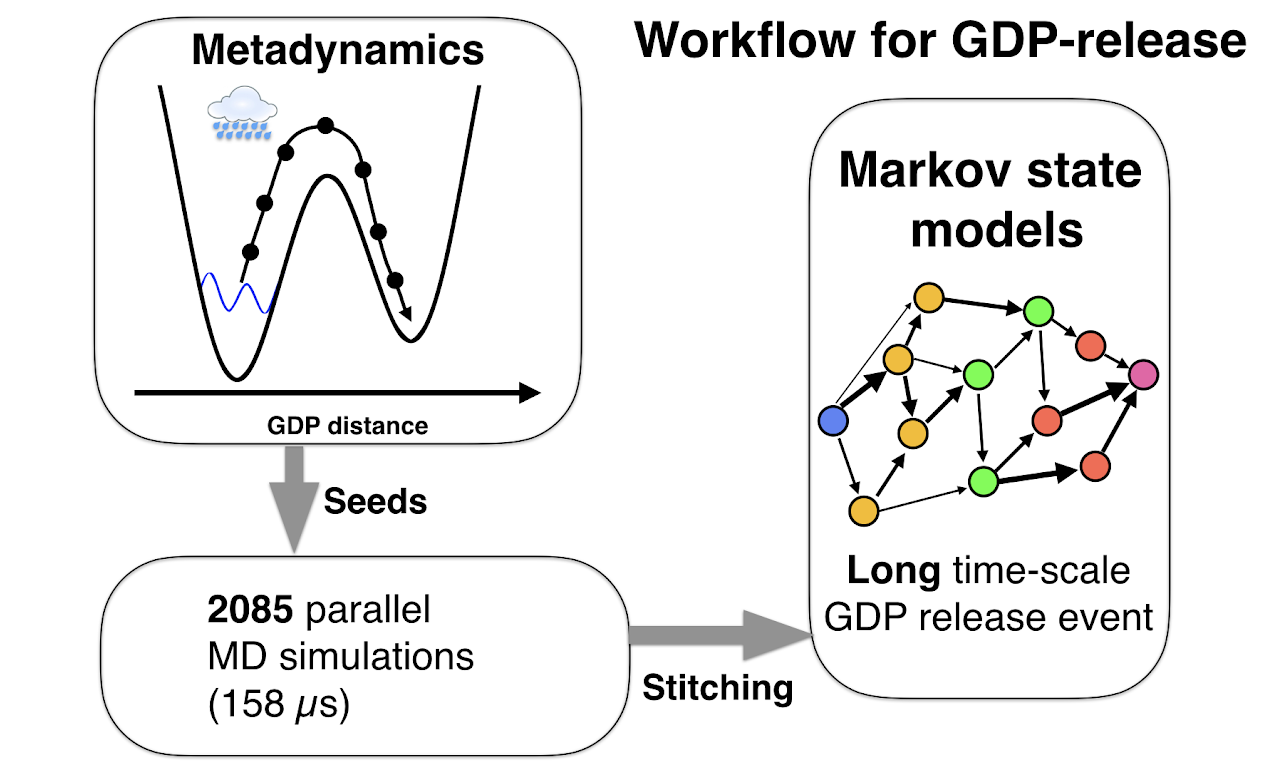
Combining powerful computational methods let us observe GDP release using unbiased simulations:
Simulations act as a “computational microscope,” capturing how Gα moves and changes shape. However, they can be computationally intensive, and so capturing a process that takes milliseconds like GDP unbinding can be difficult. In fact, GDP release is such a slow process that modeling it has never been done before!
By combining a myriad of computational approaches, we were able to model GDP release. We used a variant of “adaptive seeding”, where we ran biased simulations using a methodology named “Metadynamics” that pushed GDP out of it’s binding pocket. These Metadynamics simulations, while observing GDP unbinding, are biased simulations, and so we cannot extract mechanistic information easily from these simulations. What we CAN do is seed unbiased simulations from these biased simulations (like in our workflow below).
This effort was made possible through the generous donations of citizen scientists through the Folding@home distributed computing platform, who volunteered to run simulations on their personal computers. Their collective contributions generated data on Gα that would have taken approximately 150 years to collect on a typical desktop computer.
Our model of GDP release unified decades of literature research:
By focusing analysis to secondary structure elements and residues that are shared across all Gα homologs, this model likely captures a universal ‘skeleton’ of changes involved in Gαq activation, expanding upon a previously proposed universal mechanism for Gαq activation. The consistency of this model with a wide variety of structural and biochemical data suggests that it is a promising foundation for future efforts to understand the determinants of GPCR-Gαq interaction specificity, how mutations cause aberrant signaling and disease, and how small molecule inhibitors modulate Gαq activation.