NOTE: I am on the job market for independent faculty positions. Reach out!
Building physics driven biomarker pairing
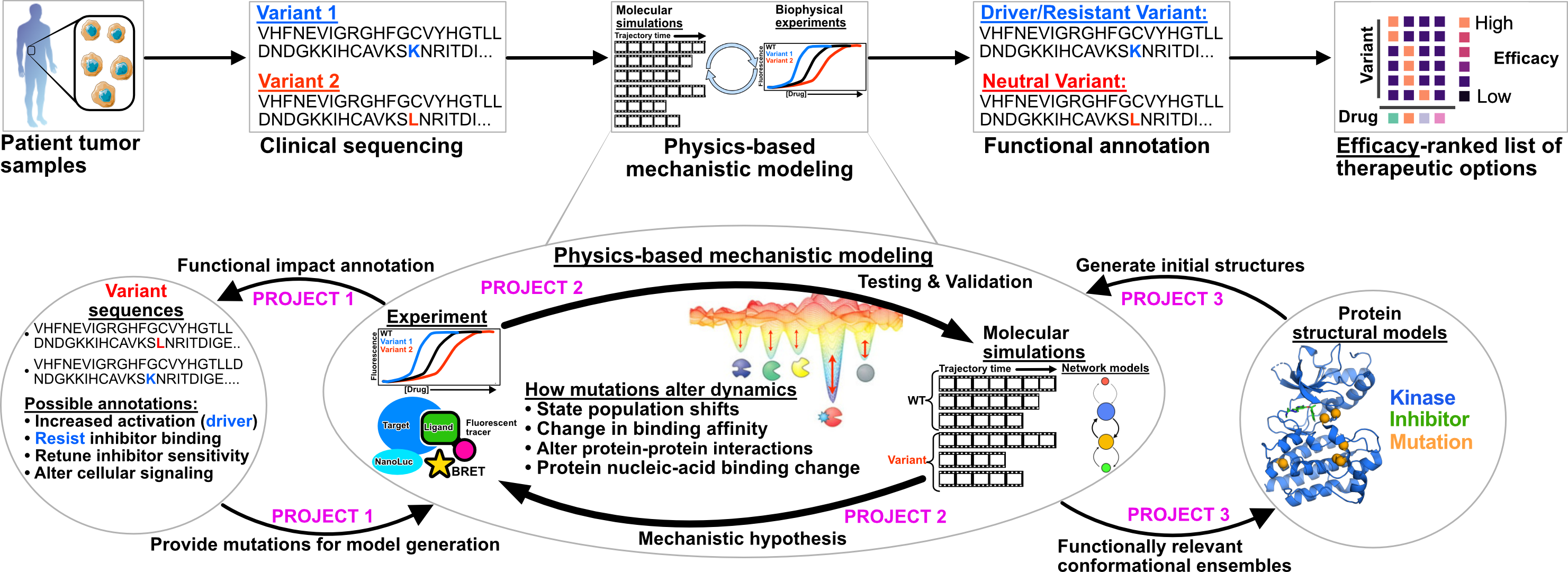
My research seeks to establish mechanism-based biomarker pairing; ranking drug options based on sensitivity, selectivity, and resistance. Rankings will derive from biophysical models built with frontier computational technologies and high throughput experiments that predict the functional impact of patient mutations.
A list of my publications can be found here. Read below to see a more details about my research interests.
Background: Individually occurring clinical variants are too rare for statistical prediction
Up to 40% of tumor diagnoses encounter therapeutic resistance in some form, spanning treatment insensitivity to compensatory mechanisms and more. Modern clinical genomics and precision oncology approaches, pairing optimally effective inhibitors with specific tumor profiles, have helped us link mutations to cancer therapy resistance.
However, across many sequencing efforts a common phenomena emerges: Individually occurring clinical variants are too infrequent to statistically infer their impact on a tumor. In other words, many mutations occur too rarely to know whether the mutation is activating, directly impacts therapy resistance and sensitivity, or has no impact.
My research focuses on bridging this gap using biophysics to provide mechanistic information describing clinically observed variants. In doing so, I want to build towards physics-based biomarker identification that will enableus to efficiently identify the appropriate treatment for a patient that may have a drug-resistant mutation.
Structure-based biophysical models promise to help predict the impact of mutations on protein function, as considering the ensemble of structures a protein adopts can provide the necessary mechanistic insight to assess the impact of mutations on function.
There are a multitude of mechanisms through which mutations can induce resistance. For example, mutations can directly alter the binding affinity of a ligand to a protein, or they can alter the conformational ensemble a protein adopts.
My research integrates frontier computational models with biophysical experiments to classify and predict impact of mutations on drug binding, protein activity, and protein conformational ensembles.
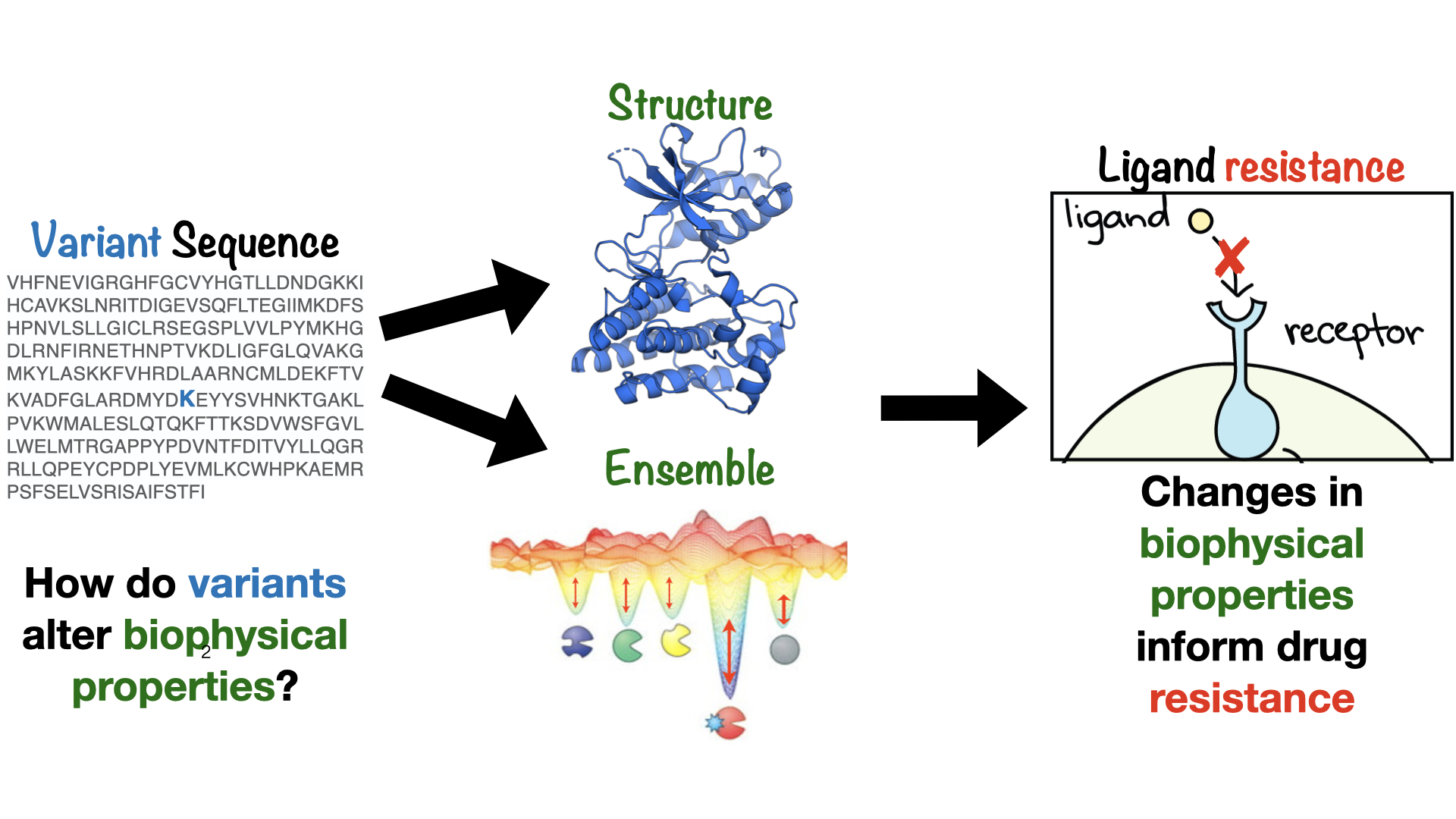
I approach this problem at three levels:
1. Identify and classify mechanisms of mutation-induced resistance and sensitivity upon mutation
(AKA What are the mechanisms through which mutations cause treatment resistance?)
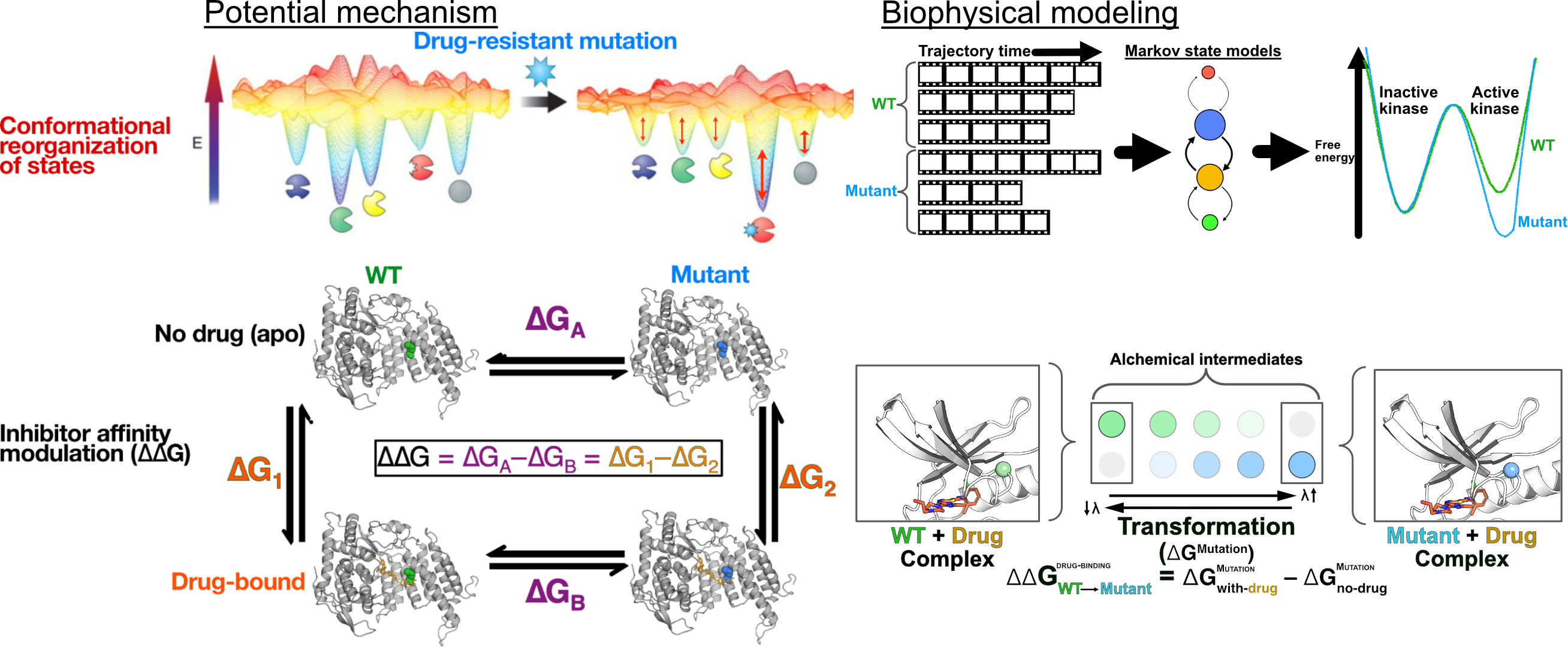
A clinical mutation may induce resistance by directly decreasing drug binding affinity, increasing protein activity, alter signaling pathways by perturbing protein-protein interactions, or retuning a target’s inhibitor sensitivity profile. I combine computation and experimental approaches to study mutation-induced mechanisms through which small-molecule inhibitor resistance may arise. This enables me to build towards models of ligand resistance, susceptibility, and selectivity.
For example: We can study the impact of mutations on ligand binding using Alchemical Free Energy calculations:
My work pairs these computational approaches with experimental validation to study how mutations may alter protein-ligand interactions. I am currently working to scale up a classic fluorescence assay from Nick Levinson’s group to exploit turn-on fluorescence to rapidly and consistently measure kinase-inhibitor binding in a label free manner. For example, the below plot shows fluorescence spectra of p38α kinase with increasing concentrations of Ponatinib, a clinically used kinase inhibitor.
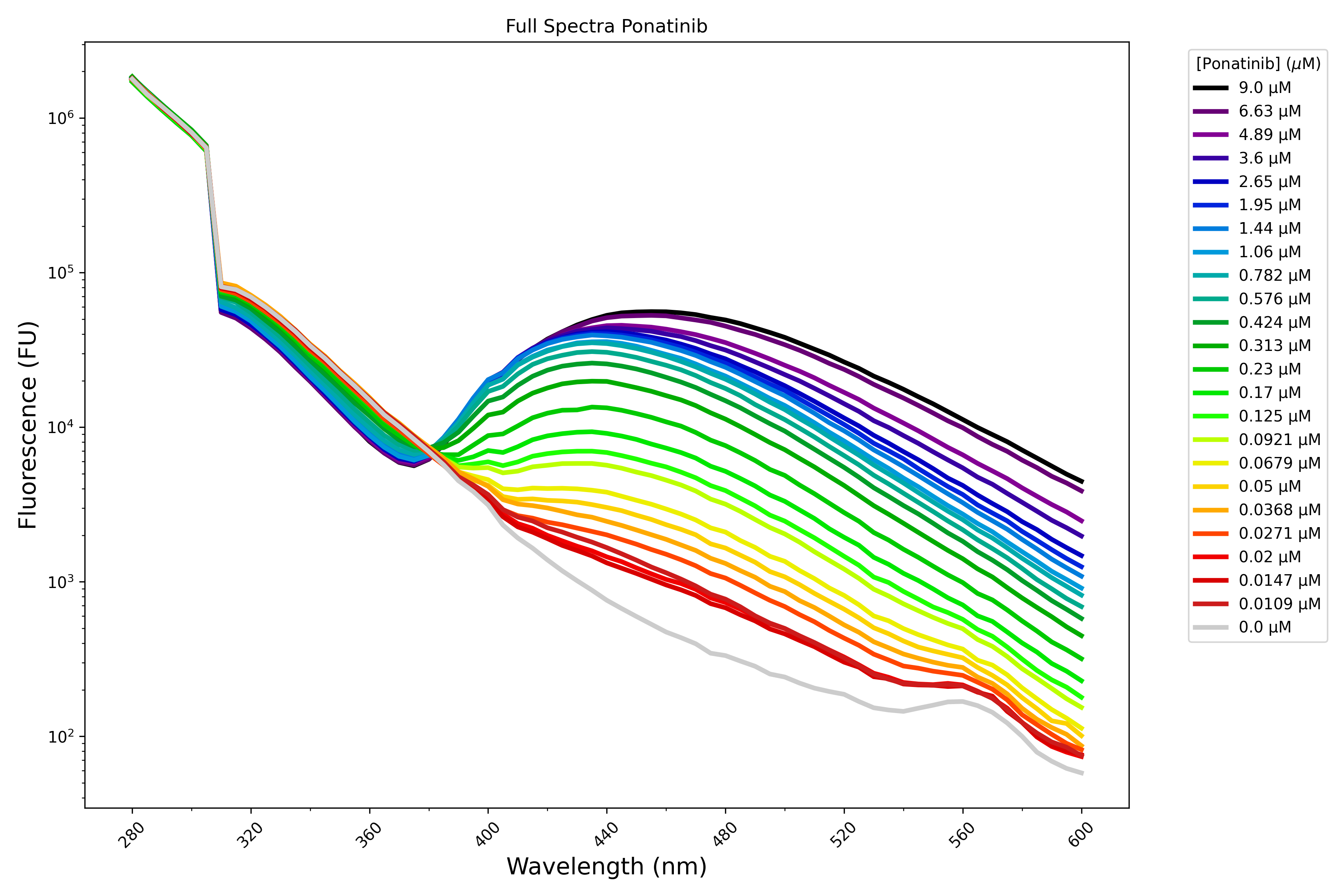
Taking slices from this emission spectra gives me the following dose response curve:
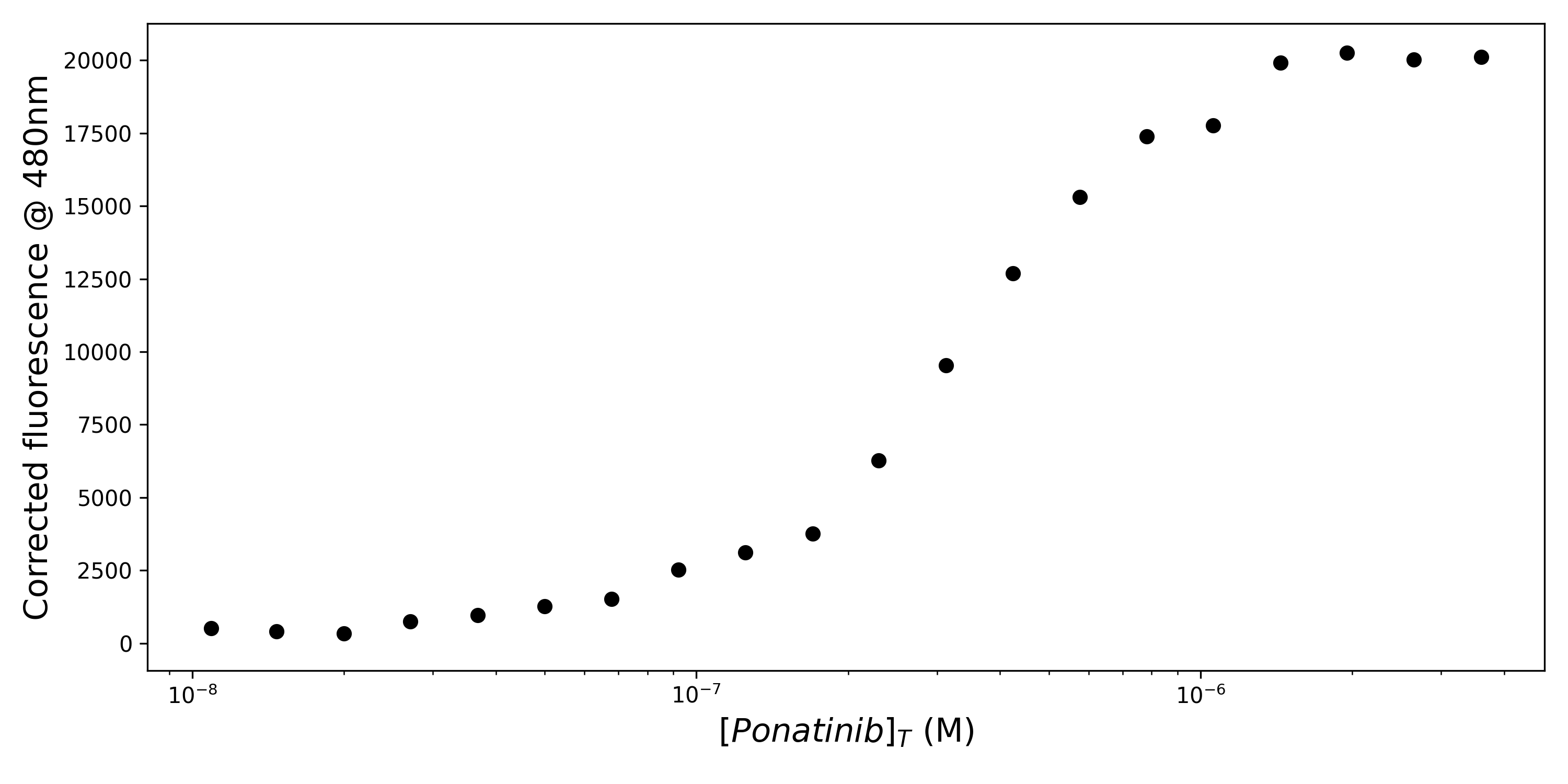
I collect these data in a high-throughput manner using liquid handlers and plate readers to rapidly assess kinase-inhibitor pairs, or variant-inhibitor pairs:

2. Predict how protein mutations alter conformational ensembles using physics-based models to alter activity
(AKA How do protein mutations alter conformational states, populations, and transitions to cause resistance?)
Mutation-induced therapy resistance is driven by changes in the conformational ensemble a protein adopts. To mechanistically model the impact of mutations on protein function, activity, and therapeutic resistance, I leverage long timescale molecular dynamics in conjunction with biophysical experimentation to study how mutations may directly impact protein ensembles.
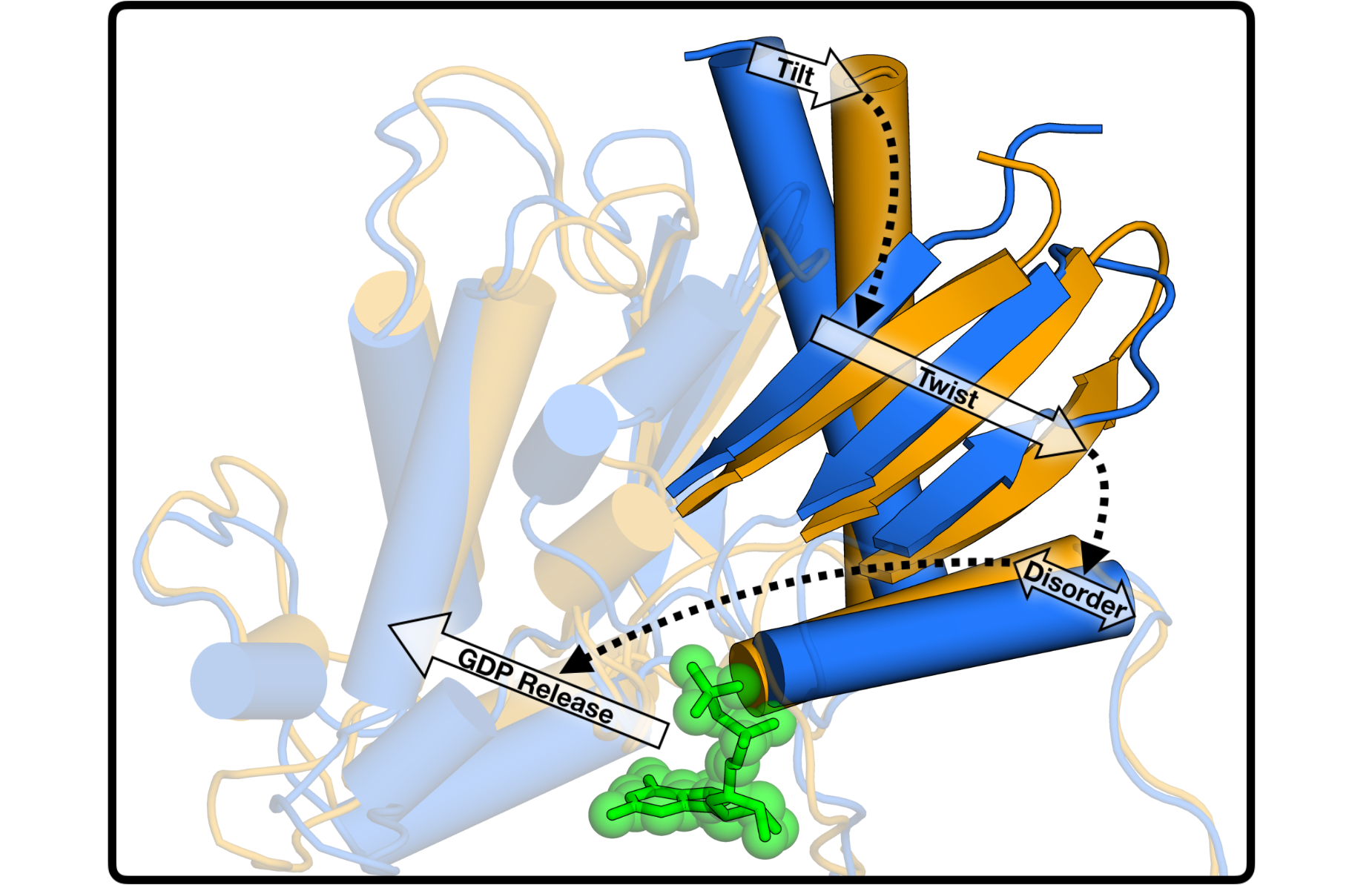
Using these tools, I seek to build models that annotate the impact of mutations on protein ensembles, and bridge the gap between biophysical modeling and phenotypic observations. This builds upon my PhD dissertation work studying mechanisms of coupling between distal regions of a protein structure (AKA allostery), which allowed us to characterize coupled events in protein activation:
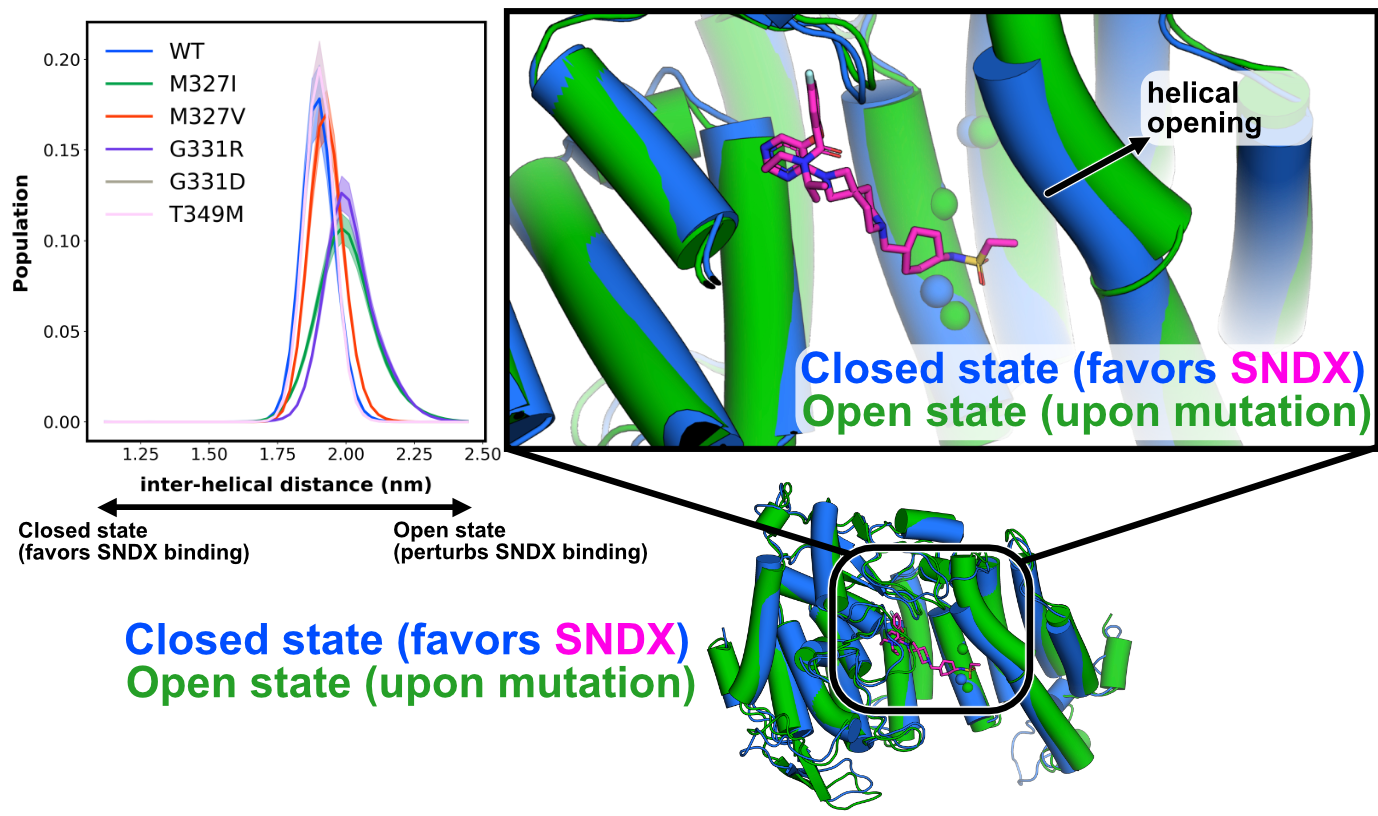
Bridging these tools with known targets, we can capture how clinical mutants shift ensembles to induce resistance, as I did so studying resistance in a Leukemia target:
3. Using distributed computing and ML-based approaches to efficiently generate and evaluate datasets describing protein ensembles
(AKA How can we efficiently generate and study ensembles of protein variants?)
Underlying my research is the need to be able to generate, analyze, and interpret protein conformational ensembles. To even collect sufficient data for ensembles can take a long time on a single GPU:
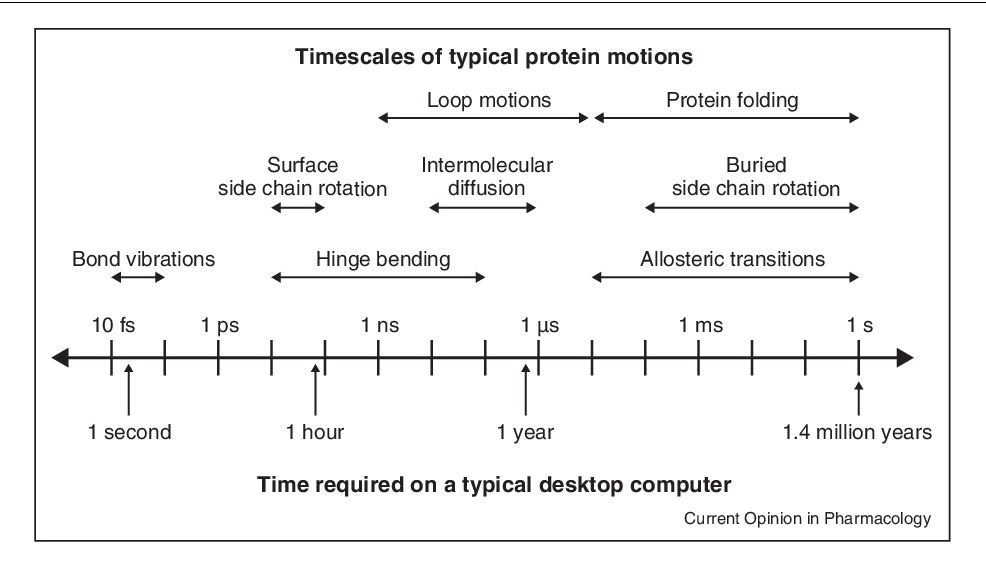
To generate these ensembles, I use long-timescale molecular simulations enabled by the Folding@home platform to generate millisecond-timescale (millions of frames) datasets of kinases and their variants.
However, generating these ensembles using distributed computing is often not sufficient compute, nor is it the more efficient use of GPU-hours. In fact, collecting milliecond-timescale simulations of kinases may not be sufficient to sample the complete set of functional kinase states.
Leveraging insights from AlphaFold and other ML-based approaches, I seek to identify principled, unbiased approaches that enable efficient exploration of the conformational landscape of many protein variants.