Chapter 6
Citizen Scientists Create an Exascale Computer to Combat COVID-19
This chapter is adapted from the following publication:
Zimmerman, M.I., Porter, J.R., Ward, M.D., Singh, S., Vithani, N., Meller, A., Mallimadugula, U.L.,
Kuhn, C.E., Borowski, J.H., Wiewiora, R.P., Hurley, M.F.D., Coffland, J.E., Voelz, V.A., Chodera, J.D.,
Bowman, G.R., Available on BiorXiv at https://doi.org/10.1101/2020.06.27.175430 [403]
My work in setting up almost all SARS-CoV-2 system for simulation and managing the Folding@home
network led figures 6.1 and 6.4 and the data described in table 6.1
6.1 Abstract
The SARS-CoV-2/COVID-19 pandemic continues to threaten global health and socioeconomic
stability. Experiments have revealed snapshots of many of the viral components but remain blind to
moving parts of these molecular machines. To capture these essential processes, over a million
citizen scientists have banded together through the Folding@home distributed computing
project to create the world’s first Exascale computer and simulate protein dynamics. An
unprecedented 0.1 seconds of simulation of the viral proteome reveal how the spike complex uses
conformational masking to evade an immune response, conformational changes implicated in
the function of other viral proteins, and ‘cryptic’ pockets that are absent in experimental
snapshots. These structures and mechanistic insights present new targets for the design of
therapeutics.
6.2 Introduction
Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) is a novel coronavirus that poses an
imminent threat to global human health and socioeconomic stability [404, 405]. With estimates of
the basic reproduction number at 3-4 and a case fatality rate for coronavirus disease 2019
(COVID-19) ranging from 0.1-12% (high temporal variation), SARS-CoV-2/COVID-19 has the
potential to spread quickly and endanger the global population [406, 405, 407, 408, 409]. As of
June 23rd, 2020, there have been over 9.1 million confirmed cases and over 472,000 fatalities,
globally. Quarantines and social distancing are effective at slowing the rate of infection; however, they
cause significant social and economic disruption. Taken together, it is crucial that we find immediate
therapeutic interventions.
A structural understanding of the SARS-CoV-2 proteins could accelerate the discovery of new
therapeutics by enabling the use of rational design [410]. Towards this end, the structural biology
community has made heroic efforts to rapidly build models of SARS-CoV-2 proteins and the
complexes they form. However, it is well established that a protein’s function is dictated by the
full range of conformations it can access; many of which remain hidden to experimental
methods. Mapping these conformations for proteins in SARS-CoV-2 will provide a clearer
picture of how they accomplish their functions, such as infecting cells, evading the immune
system, and replicating. Such maps may also present new therapeutic opportunities, such as
‘cryptic’ pockets that are absent in experimental snapshots but provide novel targets for drug
discovery.
Molecular dynamics simulations have the ability to capture the full ensemble of structures a protein
adopts but require significant computational resources. Such simulations capture an all-atom
representation of the range of motions a protein undergoes. Modern datasets often consist of a few
s of
simulation for a single protein, with a few noteworthy examples reaching ms timescales. However,
many important processes occur on slower timescales. Moreover, simulating every protein that is
relevant to SARS-CoV-2 for biologically relevant timescales would require compute resources on an
unprecedented scale.
To overcome this challenge, more than a million citizen scientists from around the world have donated
their computer resources to simulate SARS-CoV-2 proteins. This massive collaboration was enabled
by the Folding@home distributed computing platform, which has crossed the Exascale computing
barrier and is now the world’s largest supercomputer. Using this resource, we constructed quantitative
maps of the structural ensembles of over two dozen proteins and complexes that pertain
to SARS-CoV-2. Together, we have run an unprecedented 0.1 s of simulation. Our data
uncover the mechanisms of conformational changes that are essential for SARS-CoV-2’s
replication cycle and reveal a multitude of new therapeutic opportunities. The data are
supported by a variety of experimental observations and have been made publicly available
(https://covid.molssi.org/) in accordance with open science principles to accelerate the discovery of
new therapeutics.
6.3 Results and discussion
6.3.1 To the Exascale and beyond!
Folding@home is a community of citizen scientists, researchers, and tech organizations that apply
their collective computational and intellectual resources to understand the role of proteins’ dynamics
in their function and dysfunction, and to aid in the design of new proteins and therapeutics. The
project was founded in the year 2000 with the intent of understanding how proteins fold. At the time,
simulating the folding of even small proteins could easily take thousands of years on a single
computer. To overcome this challenge, the scientific team created a way to break these
intractable problems into small pieces that could be performed completely independently of one
another. They then created the Folding@home project to enable anyone with a computer and
an internet connection to volunteer to run these small chunks of simulation, called work
units.
Over the years, Folding@home has generalized to address many aspects of protein dynamics, and the
algorithms have developed significantly. The project has provided insight into diverse topics, ranging
from signaling mechanisms [411, 412, 48] to the connection between phenotype and
genotype [9, 59, 10]. Translational applications have included new means to combat antimicrobial
resistance, Ebola virus, and SFTS virus [49, 413, 54, 414].
In response to the COVID-19 pandemic, Folding@home quickly pivoted to focus on SARS-CoV-2
and the host factors it interacts with. Many people found the opportunity to take action at a time when
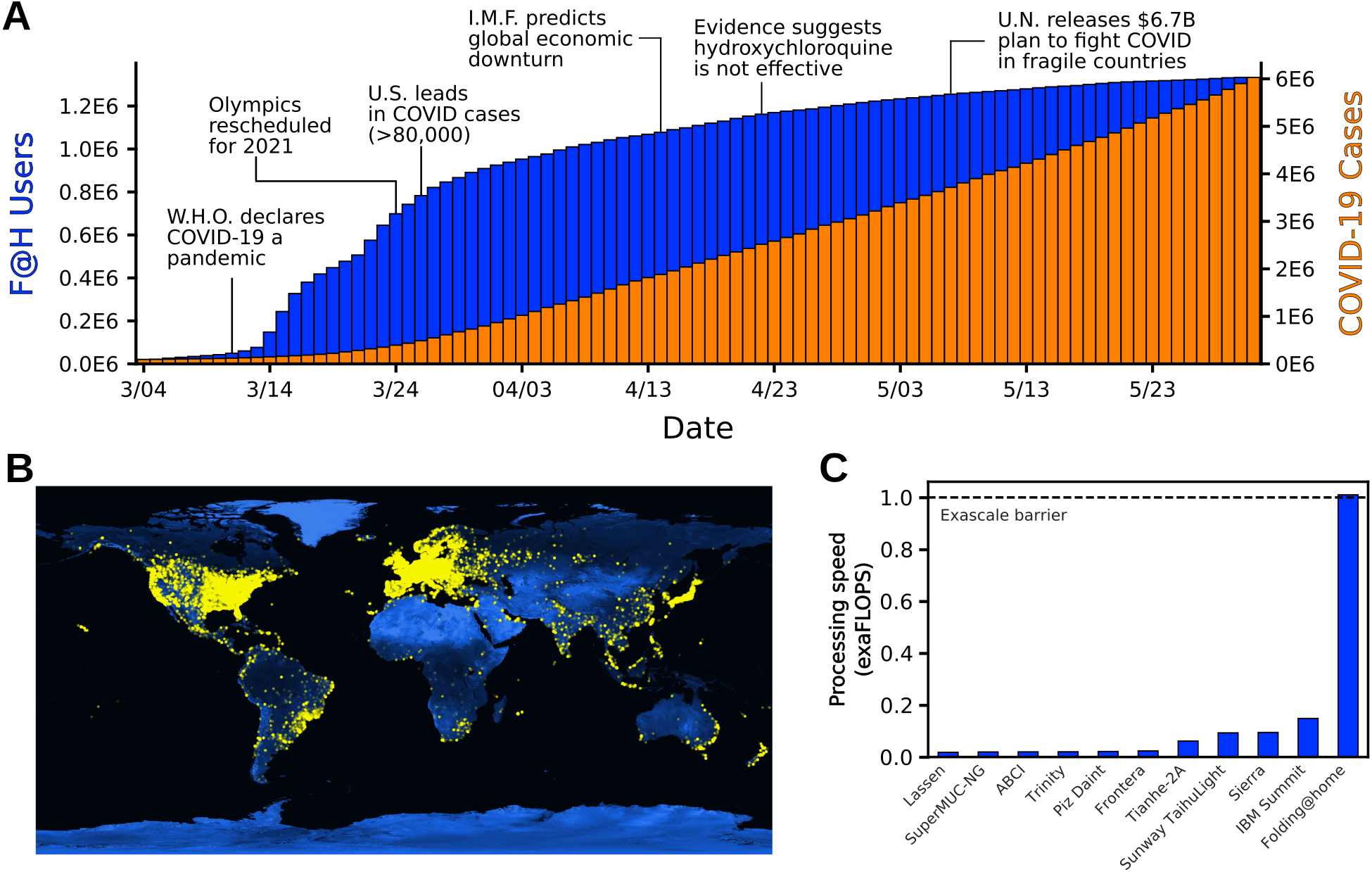
they were otherwise feeling helpless alluring. In less than three months, the project grew
from 30,000 active devices to over a million devices around the globe (Fig. 6.1A and
6.1B).
Estimating the aggregate compute power of Folding@home is non-trivial due to factors like hardware
heterogeneity, measures to maintain volunteers’ anonymity, and the fact that volunteers can turn their
machines on and off at-will. Furthermore, volunteers’ machines only communicate with the
Folding@home servers at the beginning and end of a work unit, each of which can take anywhere
from tens of minutes to a few days depending on the volunteer’s hardware and the protein to
simulate. Therefore, we chose to estimate the performance by counting the number of GPUs
and CPUs that participated in Folding@home during a three-day window and making a
conservative assumption about the computational performance of each device (see Methods for
details).
Given the above, we estimate the peak performance of Folding@home hit 1.01 exaFLOPS. This
performance was achieved at a point when 280,000 GPUs and 4.8 million CPU cores were
performing simulations. For reference, that performance is 5-fold greater than the peak performance of
the world’s fastest traditional supercomputer, called Summit (Fig. 6.1C). It is also more than the top
100 supercomputers combined. Prior to Folding@home, the first exascale supercomputer was not
scheduled to come online until the end of 2021.
6.3.2 Unmasking the spike complex
The spike complex (S) is a prominent vaccine target that is known to undergo substantial
conformational changes as part of its function [415, 416, 417]. Structurally, S is composed of three
interlocking proteins, with each chain having a cleavage site separating an S1 and S2 fragment. S
resides on the virion surface, where it waits to engage with an angiotensin-converting enzyme 2
(ACE2) receptor on a host cell to trigger infection [418, 419]. The fact that S is exposed on the virion
surface makes it an appealing vaccine target. However, it has a number of effective defense strategies.
First, S is decorated extensively with glycans that aid in immune evasion by shielding potential
antigens [420]. S also uses a conformational masking strategy, wherein it predominantly adopts a
closed conformation that buries the receptor-binding domains (RBDs) to evade immune surveillance
mechanisms. To engage with ACE2, S undergoes rare transitions to an open state that exposes the
conserved binding interface of the RBDs. Characterization of the full range of this motion is
important for understanding pathogenesis and could provide insights into novel therapeutic
options.
To capture S opening, we employed our goal-oriented adaptive sampling algorithm, FAST, in
conjunction with Folding@home. The FAST method iterates between running a batch of simulations,
building an MSM, ranking the MSM states based on how likely starting a new simulation from that
state is to yield useful data, and starting a new batch of simulations from the top ranked
states [35, 421]. The ranking function is designed to balance between favoring structures with a
desired geometric feature (in this case opening of S) and broad exploration of conformational
space. By balancing exploration-exploitation tradeoffs, FAST often captures conformational
changes with orders of magnitude less simulation time than alternative methods. Broadly
distributed structures from our FAST simulations were then used as starting points for extensive
Folding@home simulations, totaling 1 ms of data, enabling us to obtain a statistically sound final
model.
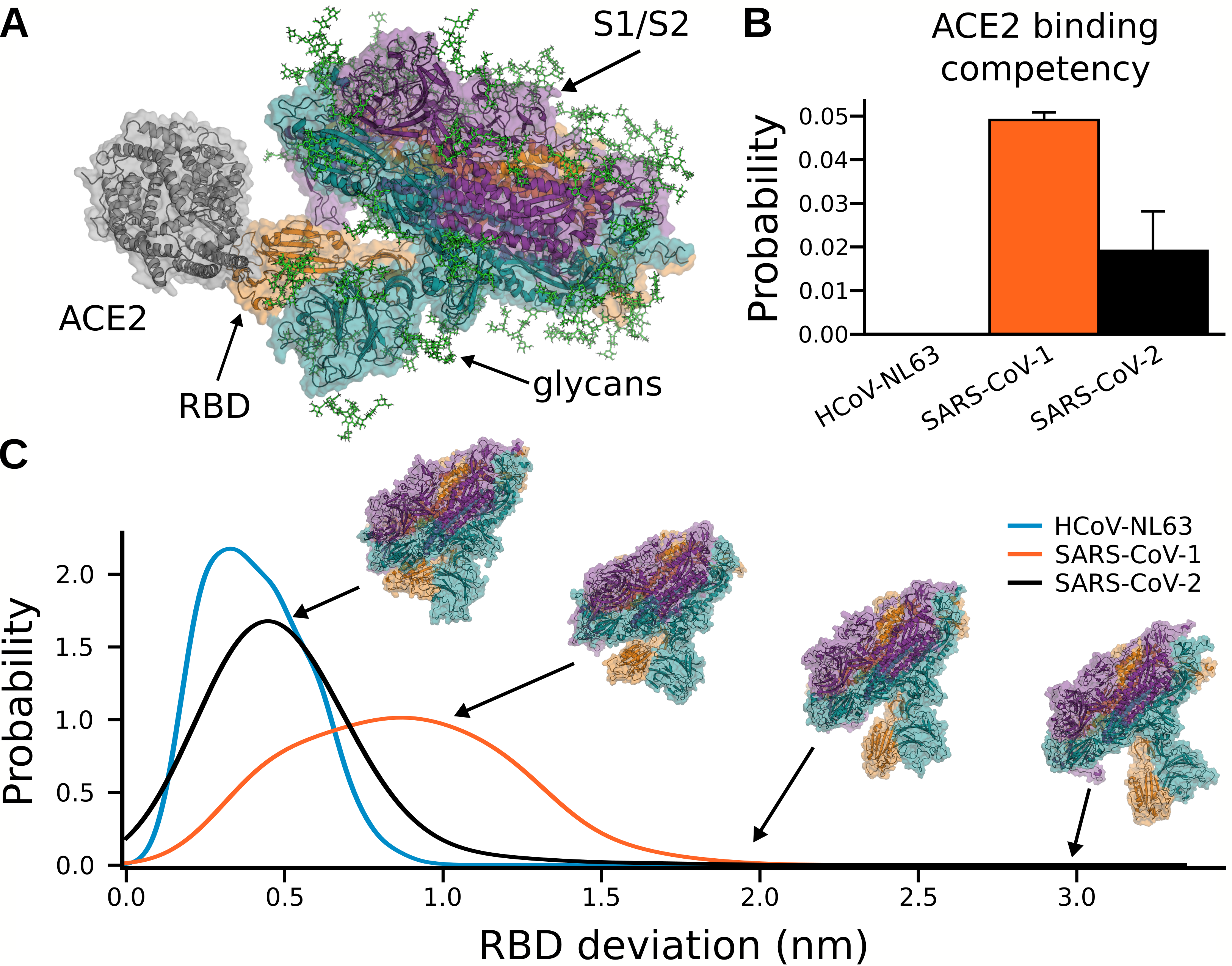
Our SARS-CoV-2 S protein simulations capture opening of S and substantial conformational
heterogeneity in the open state (Fig. 6.3). Capturing opening of S is an impressive technical feat given
that previous large-scale simulations were unable to observe this essential event for the initiation of
infection. Intriguingly, we find that opening occurs only for a single RBD at a time, akin to that
observed in cryoEM structures [422]. Additionally, we find that the scale of this opening can be
substantially larger than has been observed in experimental snapshots (Fig. 6.3). The dramatic
opening we observe is consistent with the observation that antibodies can bind to regions
of the RBD that are deeply buried and seemingly inaccessible in existing experimental
snapshots [423].
To understand the potential role of conformational masking in determining the lethality and infectivity
of different coronaviruses, we also simulated the opening of S proteins from two related viruses:
SARS-CoV-1 and HCoV-NL63. These viruses were selected because they also bind the ACE2
receptor but are associated with varying mortality rates. SARS-CoV-1 caused an outbreak in 2003
with a high case fatality rate but has not become a pandemic [424]. NL63 was discovered the
following year and continues to spread around the globe, although it is significantly less lethal
than either SARS virus [425]. We hypothesized that these phenotypic differences may be
explained by changes to the S conformational ensemble. Specifically, we propose mutations or
other perturbations can increase the S-ACE2 affinity by increasing the probability that S
adopts an open conformation or by increasing the affinity between an exposed RBD and
ACE2.
As expected, the three S complexes have very different propensities to adopt an open state and bind
ACE2. Structures from each ensemble were classified as competent to bind ACE2 if superimposing an
ACE2-RBD structure on S did not result in any steric clashes between ACE2 and the rest of the S
complex. We find that SARS-CoV-1 has the highest population of conformations that can
bind to ACE2 without steric clashes, followed by SARS-CoV-2, while opening of NL63 is
sufficiently rare that we did not observe ACE2-binding competent conformations in our
simulations (Fig. 6.2B). Interestingly, S proteins that are more likely to adopt structures that
are competent to bind ACE2 are also more likely to adopt highly open structures (Fig.
6.2C).
We also observe a number of interesting correlations between conformational masking, lethality, and
infectivity of different coronaviruses. First, more deadly coronaviruses have S proteins with less
conformational masking. Second, there is an inverse correlation between S opening and the affinity of
an isolated RBD for ACE2 (RBD-ACE2 affinities of 35 nM, 44 nM, and 185 nM for HCoV-NL63,
SARS-CoV-2, and SARS-CoV-1, respectively) [426, 427].
These observations suggest a tradeoff wherein greater conformational masking enables immune
evasion but requires a higher affinity between an exposed RBD and ACE2 to successfully infect a host
cell. We propose that the NL63 S complex is probably best at evading immune detection but is not as
infectious as the SARS viruses because strong conformational masking reduces the overall
affinity for ACE2. In contrast, the SARS-CoV-1 S complex adopts open conformations more
readily but is also more readily detected by immune surveillance mechanisms. Finally,
SARS-CoV-2 balances conformational masking and the RBD-ACE2 affinity in a manner
that allows it to evade an immune response while maintaining its ability to infect a host
cell. Based on this model, we predict that mutations that increase the probability that the
SARS-CoV-2 S complex adopts open conformations may be more lethal but spread less
readily.
Our atomically detailed model of S can enable rapid structure-based vaccine antigen design through
identification of regions minimally protected by conformational masking or the glycan shield [428].
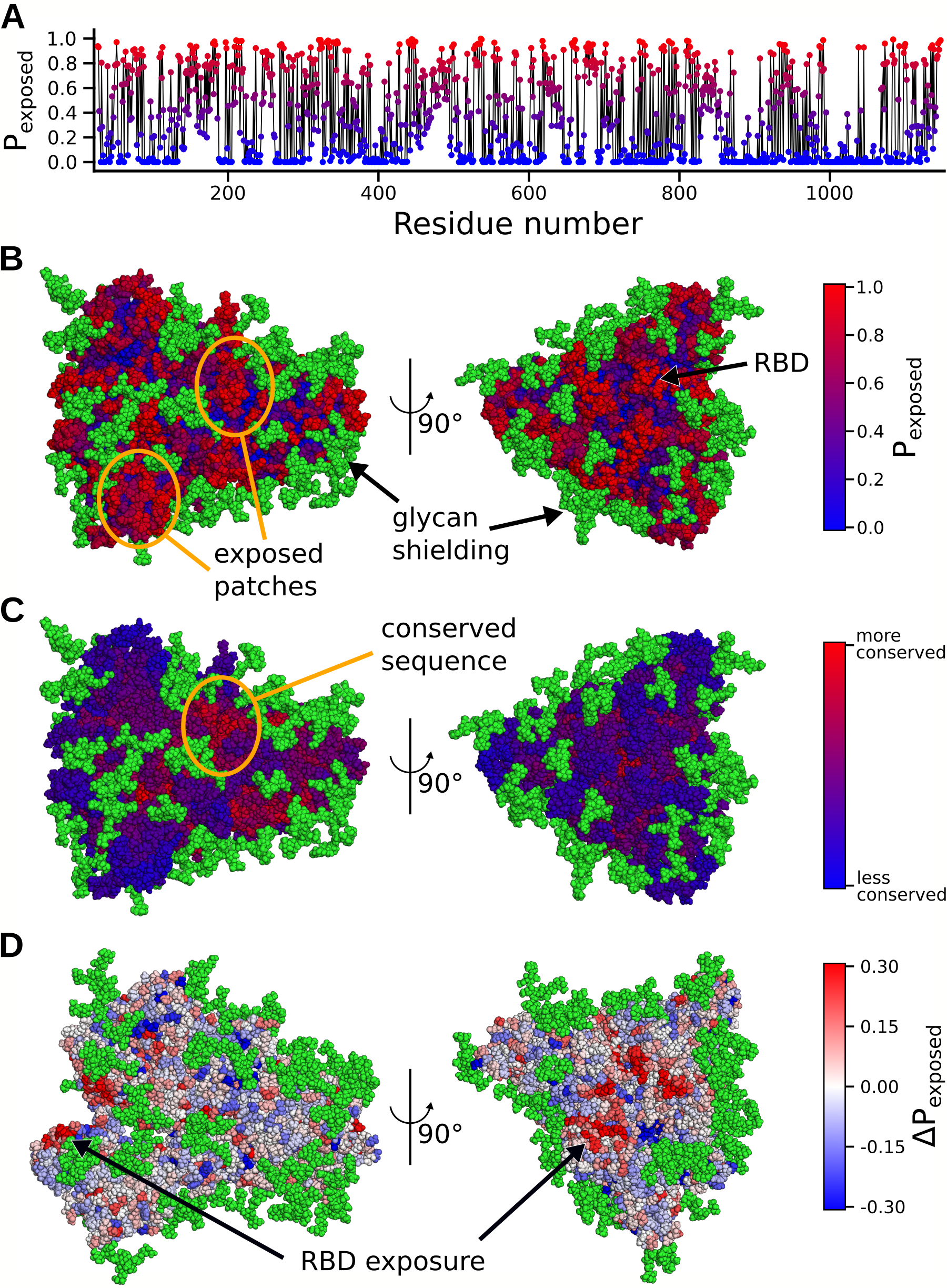
To identify these potential epitopes, we calculated the probability that each residue in S could be
exposed to therapeutics (e.g. not shielded by a glycan or buried by conformational masking), as shown
in Fig. 6.3A. Visualizing these values on the protein reveals a few patches of protein surface that
are exposed through the glycan shielding (Fig. 6.3B). However, another important factor
when targeting an antigen is picking a region with a conserved sequence to yield broader
and longer lasting efficacy. Not surprisingly, many of the exposed regions do not have a
strongly conserved sequence. Promisingly, though, we do find a conserved area with a larger
degree of solvent exposure (Fig. 6.3C). Another possibility for antigen design is to exploit
the opening motion. A number of conserved residues of the RBD show an increase in
exposure by 3̃0% in ACE2 binding competent structures (Fig. 6.3C). Consistent with
immunoassays, this region was recently found to be a cluster for neutralizing antibody
binding [423, 424].
6.3.3 Cryptic pockets and functional dynamics
Every protein in SARS-CoV-2 is a potential drug target. So, to understand their role in disease and
help progress the design of antivirals, we unleashed the full power of Folding@home to simulate
dozens of systems related to pathogenesis. While we are interested in all aspects of a proteins’
functional dynamics, expanding on the number of antiviral targets is of immediate value. Towards this
end, we seeded Folding@home simulations from our FAST-pockets adaptive sampling to aid in
the discovery of cryptic pockets. We briefly discuss two illustrative examples, out of 36
datasets.
Nonstructural protein number 5 (NSP5, also named the main protease, Mpro, or
3CLpro) is critical for the lifecycle of coronaviruses and is a major target for the design
of antivirals [429]. It is highly conserved between coronaviruses, owing to its necessary
function of processing polyproteins. NSP5 is only active as a dimer, however it exists in
a monomer-dimer equilibrium with estimates of its dissociation constant in the low
M
range [430]. Small molecules targeting this protein to inhibit enzymatic activity, either by altering its
active site or favoring the inactive monomer state, would be promising broad-spectrum antiviral
candidates [431].
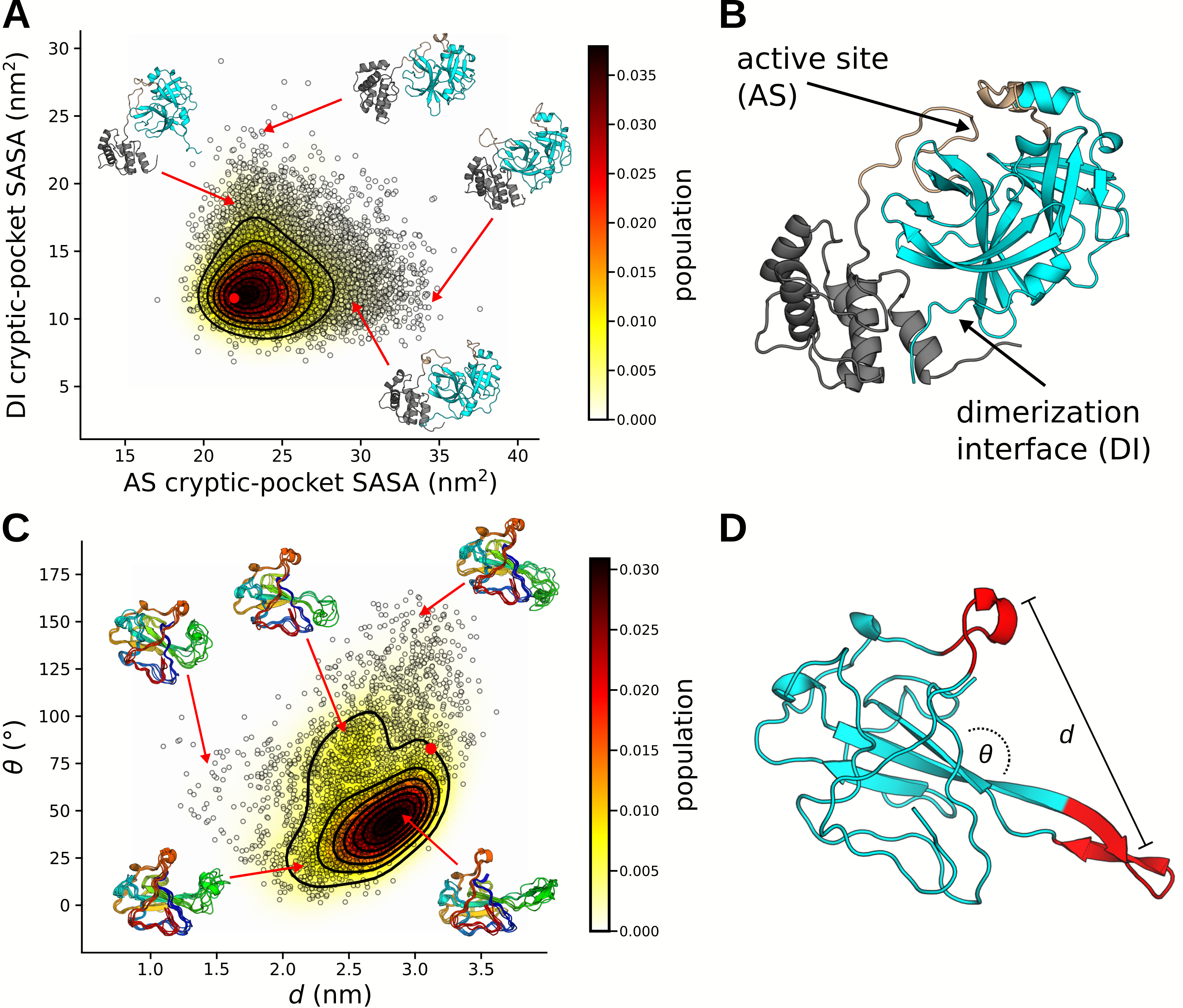
Our simulations reveal two novel cryptic pockets on NSP5 that expand our current
therapeutic options. These are shown in figure 6.4A, which projects states from our MSM
onto the solvent exposure of residues that make up the pockets. The first cryptic pocket
is an expansion of NSP5’s catalytic site. We find that the loop bridging domains II and
III is highly dynamic and can fully disengage the protein. This motion may be necessary
for catalysis and is similar to motions we have observed previously for the enzyme
-lactamase [49].
Owing to its location, a small molecule bound in this pocket is likely to prevent catalysis by
obstructing polypeptide association with catalytic residues. The second pocket is a large opening
between domains I/II and domain III. Located at the dimerization interface, this pocket offers the
possibility to find small molecule or peptide stabilizers that favor the inactive monomer
state.
In addition to cryptic pockets, our data captures many potentially functionally relevant motions within
the SARS-CoV-2 proteome. We illustrate this with the SARS-CoV-2 nucleoprotein. The
nucleoprotein is a multifunctional protein responsible for major lifecycle events such as
viral packaging, transcription, and physically linking RNA to the envelope [301, 289]. As
such, we expect the protein to accomplish these goals through a highly dynamic and rich
conformational ensemble, akin to context-dependent regulatory modules observed in Ebola virus
nucleoprotein [2, 3]. Investigating the RNA-binding domain, we observe both cryptic pockets and
an incredibly dynamic beta-hairpin, which hosts the RNA binding site, referred to as a
“positive finger” (Fig. 6.4C-D). Our observed conformational heterogeneity of the positive
finger is consistent with a structural ensemble determined using solution-state nuclear
magnetic resonance spectroscopy [432]. Our simulations also capture numerous states of the
putative RNA binding pose, where the positive finger curls up to form a cradle for RNA.
These states can provide a structural basis for the design of small molecules that would
compete with RNA binding, preventing viral assembly. Additionally, knowledge of these
probabilities can provide further insight into the mechanisms and regulation of genome
compaction/release.
The data we present in this paper represents the single largest collection of all-atom simulations. Table
6.1 is a comprehensive list of the systems we have simulated. Systems span various oligomerization
states, include important complexes, and include representation from multiple coronaviruses. We also
include human proteins that are targets for supportive therapies and preventative treatments. To
accelerate the discovery of new therapeutics and promote open science, we are posting all of our data
online (https://covid.molssi.org/).
Table 6.1: A list of protein systems we have simulated on Folding@home. Systems are
organized by viral strain and include name, oligomerization state, starting structure, number of
residues, number of atoms in the system, aggregate simulation time, and the number of cryptic
pockets we have identified.
*Missing residues were modeled using Swiss model [1].
**Structural model was generated from a homologous sequence using Swiss model [1].
***Missing residues were modeled using CHARMM-GUI [2, 3]
|
|
|
|
|
|
|
| System name | Oligomerization | Initial
structure
(homology) | Residues | Atoms in system | Aggregate simulation time (us) | Cryptic
pockets
discovered |
|
|
|
|
|
|
|
| | | | | | | |
|
|
|
|
|
|
|
| SARS-CoV-2 | | | | | | |
|
|
|
|
|
|
|
| NSP3 (Macrodomain "X") | Monomer | 6W02 | 167 | 23907 | 10,906 | - |
|
|
|
|
|
|
|
| NSP3 (Papain-like protease 2, PL2pro) | Monomer | 3E9S** | 306 | 97285 | 731 | 2 |
|
|
|
|
|
|
|
| NSP5 (main protease, 3CLpro) | Monomer | 6Y2E | 306 | 64791 | 6,405 | 2 |
|
|
|
|
|
|
|
| NSP5 (main protease, 3CLpro) | Dimer | 6Y2E | 612 | 77331 | 2,902 | 2 |
|
|
|
|
|
|
|
| NSP7 | Monomer | 5F22** | 79 | 20094 | 3,722 | 3 |
|
|
|
|
|
|
|
| NSP8 | Monomer | 2AHM** | 191 | 156282 | 1,776 | 3 |
|
|
|
|
|
|
|
| NSP9 | Dimer | 6W4B* | 226 | 49885 | 8,939 | 2 |
|
|
|
|
|
|
|
| NSP10 | Monomer | 6W4H* | 131 | 29560 | 6,141 | 2 |
|
|
|
|
|
|
|
| NSP12 (polymerase) | Monomer | 6NUR** | 891 | 186622 | 3,330 | 3 |
|
|
|
|
|
|
|
| NSP13 (helicase) | Monomer | 6JYT** | 596 | 129368 | 3,407 | 3 |
|
|
|
|
|
|
|
| NSP14 | Monomer | 5C8S** | 527 | 216380 | 2,384 | 2 |
|
|
|
|
|
|
|
| NSP15 | Monomer | 6VWW | 347 | 67345 | 3,674 | 4 |
|
|
|
|
|
|
|
| NSP15 | Hexamer | 6VWW | 2082 | 230339 | 4,270 | - |
|
|
|
|
|
|
|
| NSP16 | Monomer | 6W4H* | 298 | 45672 | 2,382 | 5 |
|
|
|
|
|
|
|
| Nucleoprotein (RBD) | Monomer | 6VYO | 173 | 29125 | 9,493 | 3 |
|
|
|
|
|
|
|
| Nucleoprotein Dimerization Domain | Monomer | 6YUN* | 118 | 34905 | 6,782 | - |
|
|
|
|
|
|
|
| Nucleoprotein Dimerization Domain | Dimer | 6YUN* | 236 | 72733 | 1,458 | 2 |
|
|
|
|
|
|
|
| Spike | Trimer | 6VXX*** | 3363 | 442881 | 1,109 | - |
|
|
|
|
|
|
|
| NSP7 / NSP8 / NSP12 | Trimer complex | 6NUR** | 1184 | 215694 | 1,686 | - |
|
|
|
|
|
|
|
| NSP10 / NSP14 | Dimer complex | 5C8S** | 688 | 226672 | 689 | 3 |
|
|
|
|
|
|
|
| NSP10 / NSP16 | Dimer complex | 6W4H* | 429 | 63752 | 3.463 | 2 |
|
|
|
|
|
|
|
| | | | | | | |
|
|
|
|
|
|
|
| SARS-CoV-1 | | | | | | |
|
|
|
|
|
|
|
| NSP3 (Macrodomain "X") | Monomer | 2FAV | 172 | 33117 | 659 | - |
|
|
|
|
|
|
|
| NSP9 | Dimer | 1QZ8* | 226 | 49599 | 7,763 | - |
|
|
|
|
|
|
|
| NSP15 | Monomer | 2H85 | 345 | 67345 | 4,734 | - |
|
|
|
|
|
|
|
| NSP15 | Hexamer | 2H85 | 2070 | 230339 | 1,130 | - |
|
|
|
|
|
|
|
| Nucleoprotein RBD | Monomer | 2OFZ | 174 | 29125 | 4,088 | - |
|
|
|
|
|
|
|
| Nucleoprotein Dimerization Domain | Monomer | 2GIB | 370 | 34905 | 1,626 | - |
|
|
|
|
|
|
|
| Nucleoprotein Dimerization Domain | Dimer | 2GIB | 740 | 72733 | 4,221 | - |
|
|
|
|
|
|
|
| Spike | Trimer | 5X58*** | 3261 | 375851 | 741 | - |
|
|
|
|
|
|
|
| NSP10 / NSP16 | Dimer complex | 6W4H** | 425 | 69589 | 518 | - |
|
|
|
|
|
|
|
| | | | | | | |
|
|
|
|
|
|
|
| Human | | | | | | |
|
|
|
|
|
|
|
| IL6 | Monomer | 1ALU | 166 | 26855 | 1,593 | 2 |
|
|
|
|
|
|
|
| IL6-R | Monomer | 1N26 | 299 | 149764 | 196 | 5 |
|
|
|
|
|
|
|
| ACE2 | Monomer | 6LZG | 596 | 75787 | 664 | 2 |
|
|
|
|
|
|
|
| | | | | | | |
|
|
|
|
|
|
|
| MERS | | | | | | |
|
|
|
|
|
|
|
| NSP13 | Monomer | 5WWP | 596 | 121134 | 719 | - |
|
|
|
|
|
|
|
| NSP10 / NSP16 | Dimer Complex | 6W4H** | 424 | 69127 | 518 | - |
|
|
|
|
|
|
|
| | | | | | | |
|
|
|
|
|
|
|
| HCoV-NL63 | | | | | | |
|
|
|
|
|
|
|
| Spike | Trimer | 5SZS*** | 3606 | 453348 | 651 | - |
|
|
|
|
|
|
|
| |
6.4 Discussion
In this work, we have utilized the largest computer in the world to tackle a global threat. The pandemic
caused by SARS-CoV-2 has necessitated a call-to-arms; a call that over a million citizen-scientists
have answered, generating more than 0.1 seconds of simulation data. The unprecedented scale of these
simulations has helped to characterize crucial stages of infection. We find that spike proteins
have a strong trade-off between making ACE2 binding interfaces accessible to infiltrate
cells and conformationally masking epitopes to subvert immune responses. SARS-CoV-2
represents a more optimal tradeoff than related coronaviruses, which may explain its success
in spreading globally. Our simulations also provide an atomically detailed roadmap for
targeting proteins for vaccines and antivirals. Furthermore, we are working on making a
comprehensive repository of cryptic pockets hosted online to accelerate the development of novel
therapeutics.
Beyond SARS-CoV-2, we expect this work to aid in a better understanding of the roles of proteins in
the coronaviridae family. Coronaviruses have been around for millennia, yet many of their proteins are
still poorly understood. Because climate change has made zoonotic transmission events more
commonplace, it is imperative that we continue to perform basic research on these viruses to better
protect us from future pandemics. For each protein system in Table 6.1, an extraordinary amount of
sampling has led to the generation of a quantitative map of its conformational landscape. There is still
much to learn about coronavirus function and these conformational ensembles contain a wealth of
information to pull from.
While we have aggressively targeted research on SARS-CoV-2, Folding@home is a general platform
for running molecular dynamics simulations at scale. Before the COVID-19 pandemic,
Folding@home was already generating datasets that were orders of magnitude greater than from
conventional means. With our explosive growth, our compute power has increased around 100-fold.
Our work here highlights the incredible utility this compute power has to rapidly understand health,
disease, and aid in drug design. Both in terms of scale and approach, we are in a new frontier of using
molecular dynamics simulations to understand biophysics; the complex task of simulating an
organism’s entire proteome could become commonplace. With the continued support of the citizen
scientists that have made this work possible, we have the opportunity to make a profound impact
on other global health crises such as cancer, neurodegenerative diseases, and antibiotic
resistance.
6.5 Methods
6.5.1 System preparations
All simulations were prepared using Gromacs 2020 [222]. Initial structures were placed in a
dodecahedron box that extends 1.0 nm beyond the protein in any dimension. Systems were then
solvated and energy minimized with a steepest descents algorithm until the maximum force fell below
100 kJ/mol/nm using a step size of 0.01 nm and a cutoff distance of 1.2 nm for the neighbor list,
Coulomb interactions, and van der Waals interactions. The AMBER03 force field was used for all
systems except spike protein with glycans, which used CHARMM36 [146, 433]. All simulations
were simulated with explicit TIP3P solvent [143].
Systems were then equilibrated for 1.0 ns, where all bonds were constrained with the LINCS
algorithm and virtual sites were used to allow a 4 fs time step [228, 226]. Cutoffs of 1.1 nm were
used for the neighbor list with 0.9 for Coulomb and van der Waals interactions. The Verlet cutoff
scheme was used for the neighbor list. The stochastic velocity rescaling (v-rescale) thermostat was
used to hold the temperature at 300 K [224].
6.5.2 Adaptive sampling simulations
The FAST algorithm was employed for each protein in Table 6.1 to enhance conformational sampling
and quickly explore dominant motions. The procedure for FAST simulations is as follows: 1) run
initial simulations, 2) build MSM, 3) rank states based on FAST ranking, 4) restart simulations from
the top ranked states, 5) repeat steps 2-4 until ranking is optimized. For each system, MSMs were
generated after each round of sampling using a k-centers clustering algorithm based on the RMSD
between select atoms. Clustering continued until the maximum distance of a frame to a cluster center
fell within a predefined cutoff. In addition to the FAST ranking, a similarity penalty was
added to promote conformational diversity in starting structures, as has been described
previously [278].
FAST-distance simulations of all Spike proteins were run at 310 K on the Microsoft Azure cloud computing
platform. The FAST-distance ranking favored states with greater RBD openings using a set of distances
between atoms. Each round of sampling was performed with 22 independent simulations that were 40 ns in
length (0.88 s
aggregate sampling per round), where the number of rounds totaled 13 (11.44
s), 22 (19.36
s), and 17
(14.96 s),
for SARS-CoV-1, SARS-CoV-2, and HCoV-NL63, respectively.
For all other proteins, FAST-pocket simulations were run at 300 K for 6 rounds,
with 10 simulations per round, where each simulation was 40 ns in length (2.4
s
aggregate simulation). The FAST-pocket ranking function favored restarting simulations from
states with large pocket openings. Pocket volumes were calculated using the LIGSITE
algorithm [434].
6.5.3 Folding@home simulations
For each adaptive sampling run, a conformationally diverse set of structures was selected to be run on
Folding@home. Structures came from the final k-centers clustering of adaptive sampling, as is
described above. Simulations were deployed using a simulation core based on either GROMACS 5.0.4
or OpenMM 7.4.1 [222, 34].
6.5.4 Markov state models
A Markov state model is a network representation of a free energy landscape and is a key tool for making
sense of molecular dynamics simulations [38]. All MSMs were built using our python package,
enspara [39]. Each system was clustered with the combined FAST and Folding@home datasets. In the
case of spike proteins, states were defined geometrically based on the RMSD between backbone
C
coordinates. States were generated as the top 3000 centers from a k-centers clustering
algorithm. All other proteins were clustered based on the euclidean distance between the
solvent accessible surface area of residues, as is described previously [49]. Systems
generated either 2500, 5000, 7500, or 10000 cluster centers from a k-centers clustering
algorithm. Select systems were refined with 1-10 k-medoid sweeps. Transition probability
matrices were produced by counting transitions between states, adding a prior count of
, and
row-normalizing, as is described previously [42]. Equilibrium populations were calculated as the
eigenvector of the transition probability matrix with an eigenvalue of one.
6.5.5 Spike/ACE2 binding competency
To determine Spike protein binding competency to ACE2 the following structures of the RBD bound
to ACE2 were used: 3D0G, 6M0J, and 3KBH, for SARS-CoV-1, SARS-CoV-2, and HCoV-NL63,
respectively. The RBD of the bound complex was superimposed onto each RBD for structures in our
MSM. Steric clashes were then determined between backbone atoms on the ACE2 molecule and the
rest of the spike protein. If any of the structures had a superposition that resulted in no clashes, it was
deemed binding competent.
6.5.6 Cryptic pockets and solvent accessible surface area
For ease of detecting cryptic pockets and other functional motions, we employed our exposon analysis
method [49]. This method correlates the solvent exposure between residues to find concerted
motions that tend to represent cryptic pocket openings. Solvent accessible surface area
calculations were computed using the Shrake-Rupley algorithm as implemented in the
python package MDTraj [148]. For all proteins and complexes, a solvent probe radius of
0.28 nm was used, which has been shown to produce a reasonable clustering and exposon
map [49].
Spike protein solvent accessible surface areas for SARS-CoV-2 were computed with glycan chains
modeled onto each cluster center. Multiple glycan rotamers were sampled for each state
and accessible surface areas for each residue were weighted based on MSM equilibrium
populations.
6.5.7 Sequence conservation
Sequence conservation of spike proteins was calculated using the Uniprot database [435]. Sequences
between 30% - 90% were pulled and aligned with the Muscle algorithm [436]. The entropy at each
position was calculated to quantify variability of amino acids. Conservation was defined as one minus
the entropy [237].
6.6 Acknowledgements
We are extremely grateful to all the citizen scientists who contributed their compute power to make
this work possible, and members of the Folding@home community who volunteered to help with
everything from technical support to translating content into multiple languages. Thanks to Microsoft
AI for Health for helping us use Azure to run adaptive sampling simulations, and to UKRI for
providing compute resources to parallelize data analysis. Thanks to Pure Storage for providing a
FlashBlade system to store our large datasets, to Seagate and Micron for additional storage, and to
MolSSI for helping organize public datasets. Thanks to Avast, AWS, Cisco, Linus Tech Tips,
Microsoft Azure, Oracle, and VMware for helping us to scale-up Folding@home’s server-side
infrastructure to keep up with the tremendous growth we experienced in such a short time. Thanks to
AMD, ARM and Neocortix, and Intel for helping to improve the performance of Folding@home on
their hardware. Thanks to all of the above companies for spreading the word about Folding@home,
and also to A16Z, Best Buy, CCP, CoreWeave, Daimler Truck AG, Dell, GitHub, HP, La
Liga, Media Monks, Microcenter, NVIDIA, and Telefonica. Thanks to CERN and the
particle physics community for providing guidance on data management strategies and to
DataDog for server monitoring services. GRB and his lab were supported by funding from
Avast, the Center for the Science and Engineering of Living Systems (CSELS), an NSF
RAPID award, NSF CAREER Award MCB-1552471, NIH R01 GM124007, a Burroughs
Wellcome Fund Career Award at the Scientific Interface, and a Packard Fellowship for
Science and Engineering. JDC acknowledges support from NIH grant P30 CA008748 and
NIH grant R01 GM121505. VAV and MFDH acknowledge support from NIH grant R01
GM123296.
6.7 Disclosures
JDC is a current member of the Scientific Advisory Board of OpenEye Scientific Software and a
consultant to Foresite Laboratories. The Chodera laboratory receives or has received funding from
multiple sources, including the National Institutes of Health, the National Science Foundation, the
Parker Institute for Cancer Immunotherapy, Relay Therapeutics, Entasis Therapeutics, Silicon
Therapeutics, EMD Serono (Merck KGaA), AstraZeneca, Vir Biotechnology, Bayer, XtalPi, the
Molecular Sciences Software Institute, the Starr Cancer Consortium, the Open Force Field
Consortium, Cycle for Survival, a Louis V. Gerstner Young Investigator Award, and the
Sloan Kettering Institute. A complete funding history for the Chodera lab can be found at
http://choderalab.org/funding.