Chapter 5
The SARS-CoV-2 nucleocapsid protein is dynamic, disordered, and phase separates with
RNA.
This chapter is adapted from the following publication:
Cubuk, J., Alston, J.J., Incicco, J.J., Singh, S., Stuchell-Brereton, M.D., Ward, M.D., Zimmerman, M.I.,
Vithani, N., Griffith, D., Wagoner, J.A., Bowman, G.R., Hall, K.B., Soranno, A., Holehouse, A.S., The
SARS-CoV-2 nucleocapsid protein is dynamic, disordered, and phase separates with RNA, Available
on Biorxiv: https://doi.org/10.1101/2020.06.17.158121 [2]
In this work, my work in setting up, simulating, and generating seed conformations of
the folded domains (and populations) led to the data presented in figures 5.1, 5.2, and
5.4.
5.1 Abstract
The SARS-CoV-2 nucleocapsid (N) protein is an abundant RNA binding protein critical for viral
genome packaging, yet the molecular details that underlie this process are poorly understood. Here we
combine single-molecule spectroscopy with all-atom simulations to uncover the molecular details that
contribute to N protein function. N protein contains three dynamic disordered regions that house
putative transiently-helical binding motifs. The two folded domains interact minimally such
that full-length N protein is a flexible and multivalent RNA binding protein. N protein
also undergoes liquid-liquid phase separation when mixed with RNA, and polymer theory
predicts that the same multivalent interactions that drive phase separation also engender RNA
compaction. We offer a simple symmetry-breaking model that provides a plausible route
through which single-genome condensation preferentially occurs over phase separation,
suggesting that phase separation offers a convenient macroscopic readout of a key nanoscopic
interaction.
5.2 Introduction
Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) is an enveloped, positive-strand RNA
virus that causes the disease COVID-19 (Coronavirus Disease-2019) [279]. While coronaviruses
typically cause relatively mild respiratory diseases, COVID-19 is on course to kill half a million
people in the first six months since its emergence in late 2019 [279, 280, 281]. Given the timeframe
for vaccine development is on the order of months to years, alternative therapeutic approaches are
sought to ameliorate viral morbidity and mortality [282].
A challenge in identifying candidate drugs is our relatively sparse understanding of the molecular
details that underlie the function of SARS-CoV-2 proteins. As a result, there is a surge of biochemical
and biophysical exploration of these proteins, with the ultimate goal of identifying proteins that are
suitable targets for disruption, ideally with insight into the molecular details of how disruption could
be achieved [283, 284].
While much attention has been focused on the Spike (S) protein, many other SARS-CoV-2 proteins
play equally critical roles in viral physiology, yet we know relatively little about their structural or
biophysical properties [285, 286, 287, 288]. Here we performed a high-resolution structural and
biophysical characterization of the SARS-CoV-2 nucleocapsid (N) protein, the protein responsible for
genome packaging [289, 290, 291]. A large fraction of N protein is predicted to be intrinsically
disordered, which constitutes a major barrier to conventional structural characterization [290]. To
overcome these limitations, we combined single-molecule spectroscopy with all-atom simulations
to build a residue-by-residue description of all three disordered regions in the context of
their folded domains The combination of single-molecule spectroscopy and simulations to
reconstruct structural ensembles has been applied extensively to uncover key molecular
details underlying disordered protein regions [292, 293, 294, 295, 296, 297]. Our goal
here is to provide biophysical and structural insights into the physical basis of N protein
function.
In exploring the molecular properties of N protein, we discovered it undergoes phase separation with
RNA, as was also reported recently [298, 299, 300]. Given N protein underlies viral packaging,
we reasoned phase separation may in fact be an unavoidable epiphenomenon that reflects
physical properties necessary to drive compaction of long RNA molecules. To explore
this principle further, we developed a simple physical model, which suggested symmetry
breaking through a small number of high-affinity binding sites can organize anisotropic
multivalent interactions to drive single-polymer compaction, as opposed to multi-polymer phase
separation. Irrespective of its physiological role, our results suggest that phase separation
provides a macroscopic readout (visible droplets) of a nanoscopic process (protein:RNA and
protein:protein interaction). In the context of SARS-CoV-2, those interactions are expected to be key
for viral packaging, such that assays which monitor phase separation of N protein with
RNA may offer a convenient route to identify compounds that will also attenuate viral
assembly.
5.3 Results
Coronavirus nucleocapsid proteins are multi-domain RNA binding proteins that play a critical role in
many aspects of the viral life cycle [291, 301]. The SARS-CoV-2 N protein shares a number of
sequence features with other nucleocapsid proteins from coronaviruses (Fig. D.1-D.5). Work on N
protein from a range of model coronaviruses has shown that N protein undergoes both
self-association, interaction with other proteins, and interaction with RNA, all in a highly multivalent
manner.
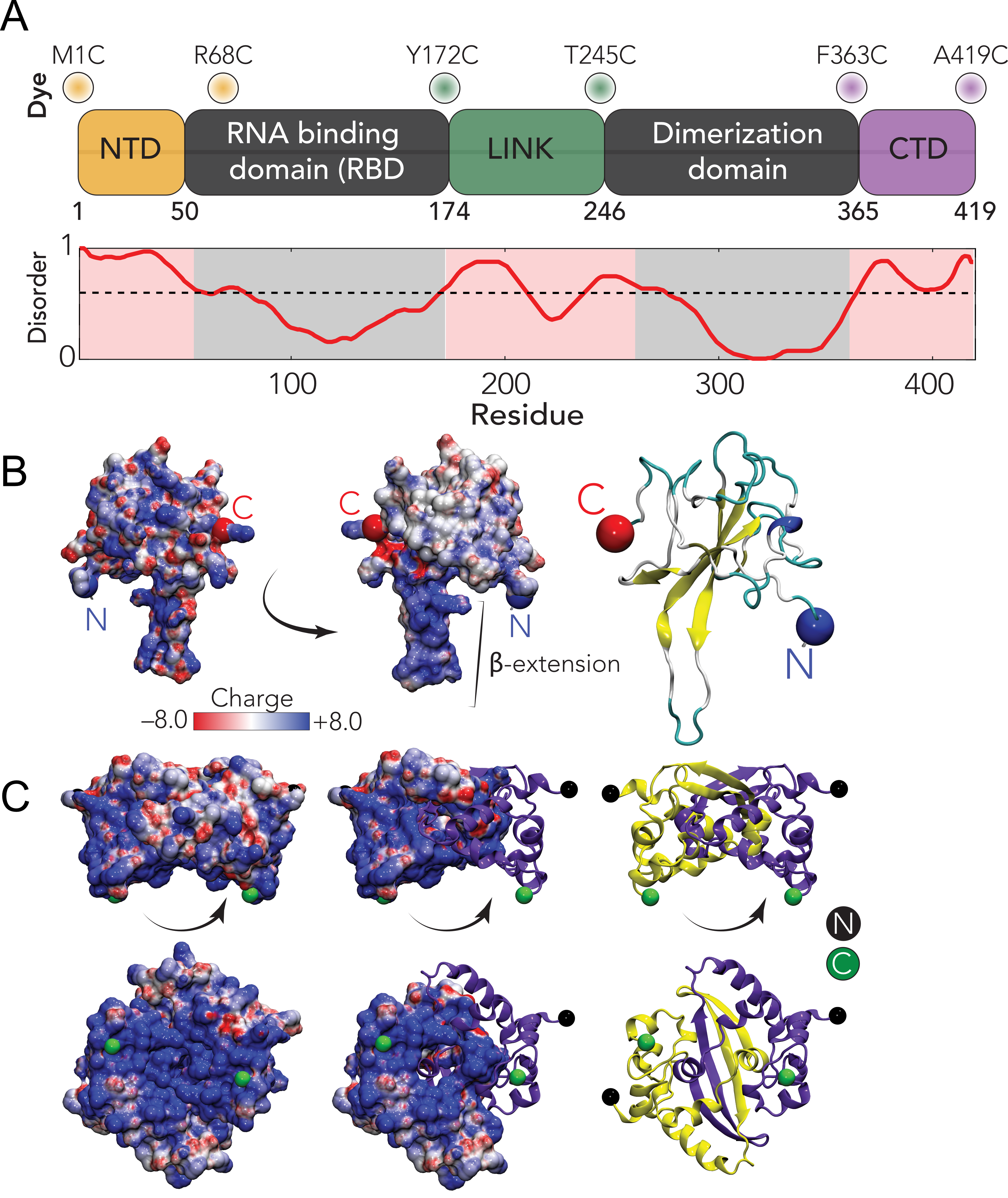
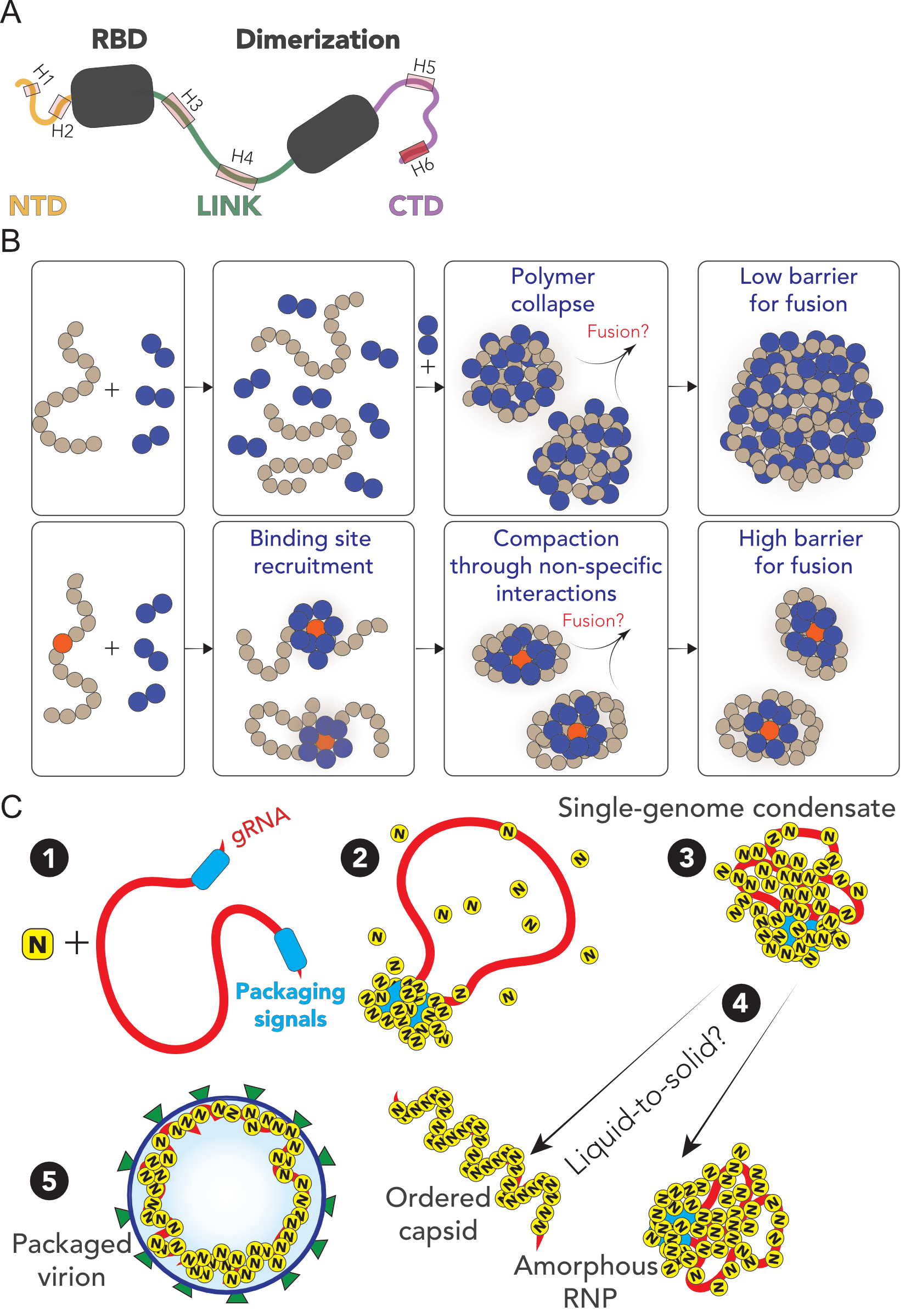
The SARS-CoV-2 N protein can be divided into five domains; a predicted intrinsically disordered
N-terminal domain (NTD), an RNA binding domain (RBD), a predicted disordered central linker
(LINK), a dimerization domain, and a predicted disordered C-terminal domain (CTD) (Fig. 5.1).
While SARS-CoV-2 is a novel coronavirus, decades of work on model coronaviruses (including SARS
coronavirus) have revealed a number of features expected to hold true in the SARS-CoV-2 N protein.
Notably, all five domains are predicted to bind RNA [303, 304, 305, 306, 307, 308, 309], and
while the dimerization domain facilitates the formation of well-defined stoichiometric dimers,
RNA-independent higher-order oligomerization is also expected to occur [308, 310, 311, 312].
Importantly, protein-protein and protein-RNA interaction sites have been mapped to all three
disordered regions.
Despite recent structures of the RBD and dimerization domains from SARS-CoV-2, the solution-state
conformational behavior of the full-length protein remains elusive [313, 314, 315]. Understanding N
protein function necessitates a mechanistic understanding of the flexible predicted disordered
regions and their interplay with the folded domains. A recent small-angle X-ray study
shows good agreement with previous work on SARS, suggesting the LINK is relatively
extended, but neither the structural basis for this extension nor the underlying dynamics are
known [303, 316].
Here, we address these questions by probing three three full-length constructs of the N protein
with fluorescent labels (Alexa 488 and 594) flanking the NTD, the LINK, and the CTD
(see Fig. 5.1A). These constructs allow us to probe conformations and dynamics of the
disordered regions in the context of the full-length protein using single-molecule Förster Resonance Energy Transfer (FRET) and Fluorescence Correlation Spectroscopy (FCS)
(see SI for details). In parallel to the experiments, we performed all-atom Monte Carlo
simulations of each of the three IDRs in isolation and in context with their adjacent folded
domains.
5.3.1 The NTD is disordered, flexible, and transiently interacts with the RBD.
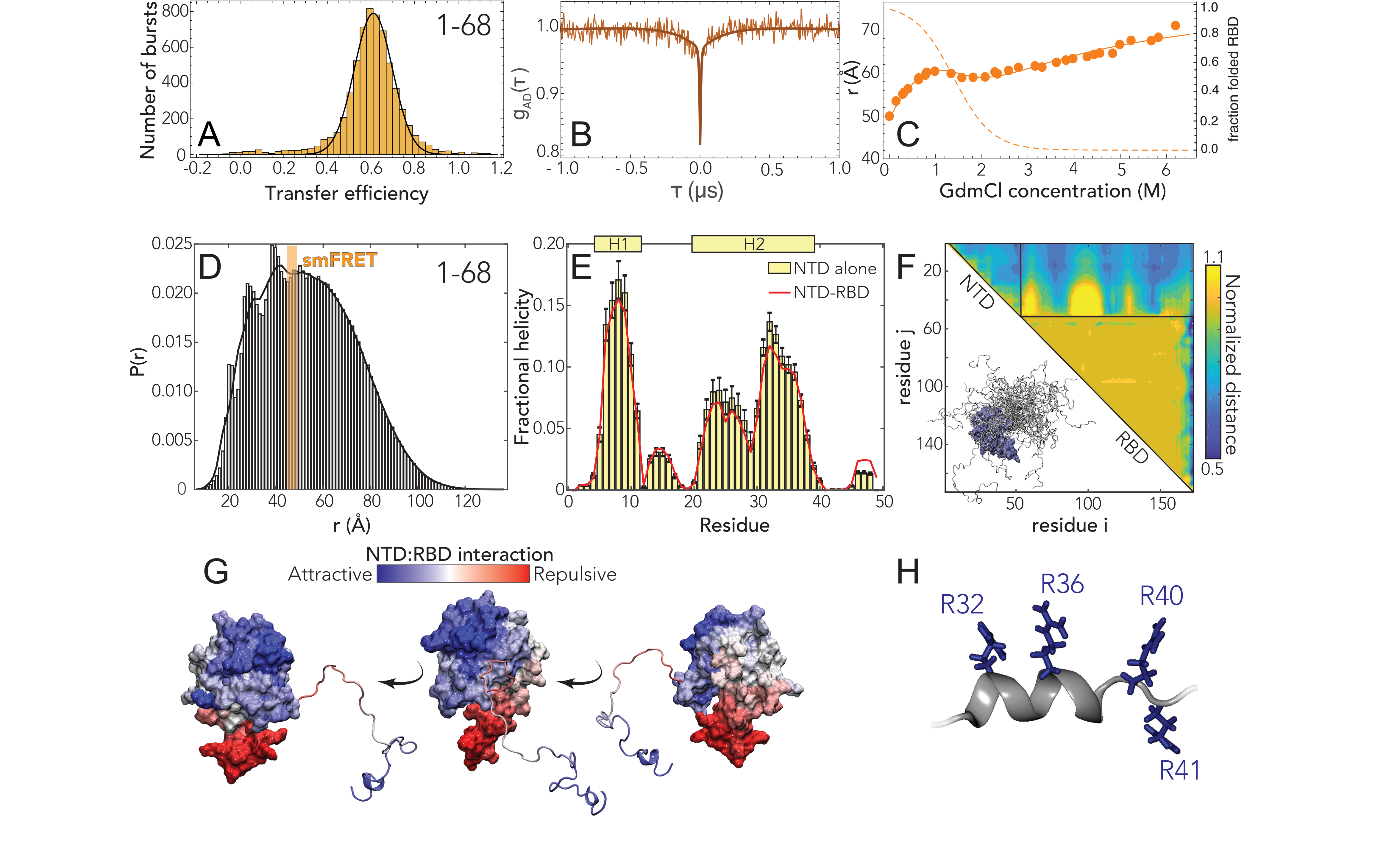
We started our analysis by investigating the NTD conformations. Under native conditions, single-molecule
FRET measurements revealed the occurrence of a single population with a mean transfer efficiency of
0.61
0.03 (Fig. 5.2A and Fig. D.6). To assess whether this transfer efficiency reports about a rigid
distance (e.g. structure formation or persistent interaction with the RBD) or is a dynamic
average across multiple conformations, we first compare the lifetime of the fluorophores with
transfer efficiency. Under native conditions, the donor and acceptor lifetimes for the NTD
construct lie on the line that represents fast conformational dynamics (Fig. D.8A). To properly
quantify the timescale associated with these fast structural rearrangements, we leveraged
nanoseconds FCS. As expected for a dynamic population [317, 318], the cross-correlation of
acceptor-donor photons for the NTD is anticorrelated (Fig. 5.2B and D.11). A global fit of the
donor-donor, acceptor-acceptor, and acceptor-donor correlations yields a reconfiguration time
= 170
30 ns.
This is longer than reconfiguration times observed for other proteins with a similar persistence length
and charge content [318, 319, 320, 321], hinting at a large contribution from internal friction due to
rapid intramolecular contacts (formed either within the NTD or with the RBD) or transient formation
of short structural motifs [322].
As a next step, we assessed the stability of the folded RBD and its influence on the conformations of
the NTD by studying the effect of a chemical denaturant on the protein. The titration with
guanidinium chloride (GdmCl) reveals a decrease of transfer efficiencies when moving from native
buffer conditions to 1 M GdmCl, followed by a plateau of the transfer efficiencies at concentrations
between 1 M and 2 M and a subsequent further decrease at higher concentrations (Fig. D.6 and D.8).
This behavior can be understood assuming that the plateau between 1 M and 2 M GdmCl represents
the average of transfer efficiencies between two populations in equilibrium that have very close
transfer efficiency and are not resolved because of shot noise. Indeed, this interpretation is supported
by a broadening in the transfer efficiency peak between 1 M and 2 M GdmCl, which is expected if two
overlapping populations react differently to denaturant. Besides the effect of the unfolding of the
RBD, the dimensions of the NTD are also modulated by change in the solvent quality when adding
denaturant (Fig. 5.2C, D.6, D.8) and this contribution to the expansion of the chain can be
described using an empirical binding model [323, 324, 325, 326, 327]. A fit of the interdye
root-mean-square distances to this model and the extracted stability of RBD (midpoint: 1.25
0.2 M;
= (3
0.6)
RT) is presented in Fig. 5.2C A comparative fit of the histograms with two populations yields an
identical result in terms of RBD stability and protein conformations (Fig. D.9).
These observations provide two important insights. Firstly, the RBD is completely folded under native
conditions (Fig. 5.2C). Secondly, the RBD contributes significantly to the conformations of the
measured NTD construct, mainly by reducing the accessible space of the disordered tail and favoring
expanded configurations, as shown by the shift in transfer efficiency when the RBD is
unfolded.
To better understand the sequence-dependent conformational behavior of the NTD we
turned to all-atom simulations of an NTD-RBD construct. We used a novel sequential
sampling approach that integrates long timescale MD simulations performed using the
Folding@home distributed computing platform with all-atom Monte Carlo simulation
performed with the ABSINTH forcefield to generate an ensemble of almost 400,000 distinct
conformations (see methods [37, 328]. We also performed simulations of the NTD in
isolation.
We observed excellent agreement between simulation and experiment for the equivalent inter-residue
distance (Fig. 5.2D). The peaks on the left side of the histogram reflect specific simulations where the
NTD engages more extensively with the RBD through a fuzzy interaction, leading to local kinetic
traps [328]. We also identified several regions in the NTD where transient helices form,
and using normalized distance maps found regions of transient attractive and repulsive
interaction between the NTD and the RBD. In particular, the basic beta-strand extension
from the RBD (Fig. 5.1B) repels the arginine-rich C-terminal region of the NTD, while a
phenylalanine residue (F17) in the NTD engages with a hydrophobic face on the RBD (Fig. 5.2G).
Finally, we noticed the arginine-rich C-terminal residues (residues 31 - 41) form a transient
alpha helix projecting three of the four arginines in the same direction (Fig. 5.2H). These
features provide molecular insight into previously reported functional observations (see
Discussion).
5.3.2 The linker is highly dynamic and there is minimal interaction between the RBD and the
dimerization domain.
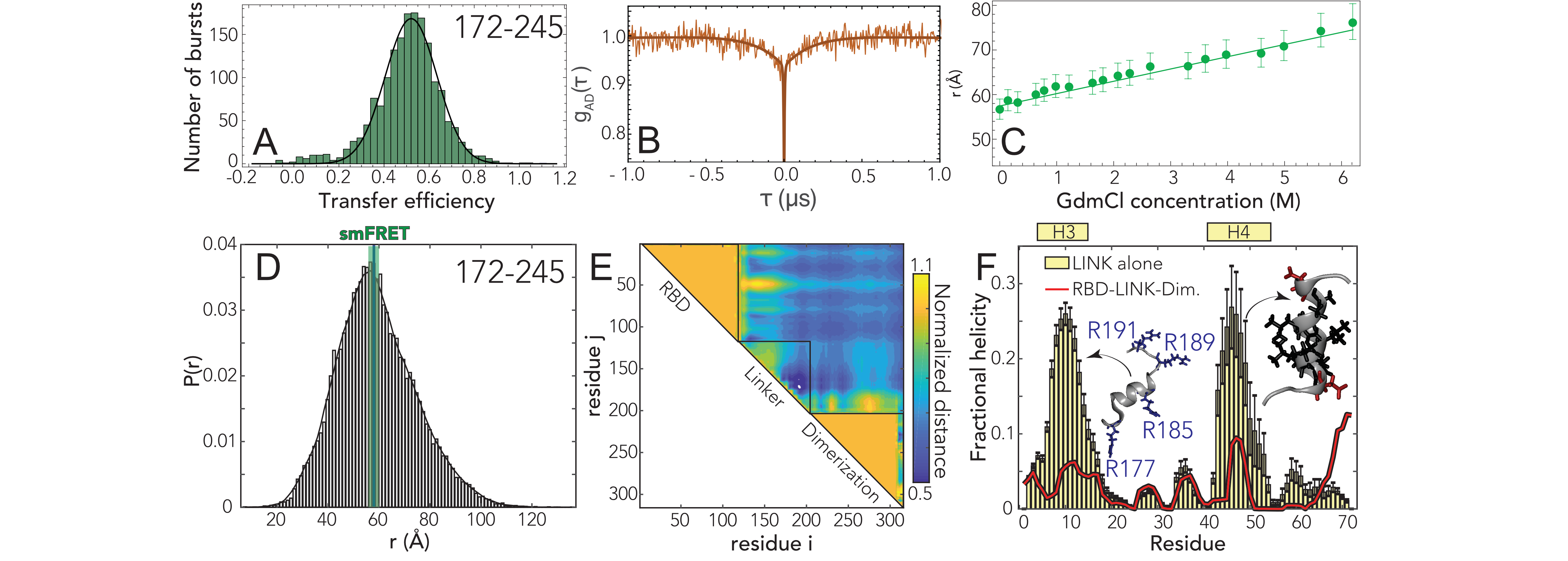
We next turned to the linker (LINK) construct to investigate how the disordered region modulates the
interaction and dynamics between the two folded domains. Under native conditions (50 mM Tris
buffer), single-molecule FRET reveals a narrow population with mean transfer efficiency of 0.52
0.03.
Comparison of the fluorescence lifetime and transfer efficiency indicates that, like the NTD, the
transfer efficiency represents a dynamic conformational ensemble sampled by the LINK
(Fig. D.7B). ns-FCS confirms fast dynamics with a characteristic reconfiguration time
of 120
20 ns
(Fig. 5.3B and D.11). This reconfiguration time is compatible with high internal friction
effects, as observed for other unstructured proteins [318, 319], but may also account for the
drag of the surrounding domains. The root-mean-square interdye distance for the LINK
is equal to
57 2 Å
(= 5.8
0.4 Å) when assuming a Gaussian Chain distribution and 55
2 Å
(= 5.4
0.4 Å)
when using a SAW model (see appendix E).
Next, we addressed whether the LINK segment populates elements of persistent secondary structure
or forms stable interaction with the RBD or dimerization domains. Addition of the denaturant shows a
continuous shift of the transfer efficiency toward lower values (Fig. D.6,D.8), that corresponds to an
almost linear expansion of the chain (see Fig. 5.3C). These observations support a model in which
LINK is unstructured and flexible and do not reveal a significant fraction of folding or persistent
interactions with or between folded domains. Overall, our single-molecule observations report a
relatively extended average inter-domain distance, suggesting a low number of interactions
between folded domains. To further explore this conclusion, we turned again to Monte Carlo
simulations.
As with the NTD, all-atom Monte Carlo simulations provide atomistic insight that can be
compared with our spectroscopic results. Given the size of the system an alternative sampling
strategy to the NTD-RBD construct was pursued here that did not include MD simulations of
the folded domains, but we instead ran simulations of a construct that included the RBD,
LINK and dimerization domain. In addition, we also performed simulations of the LINK in
isolation.
We again found good agreement between simulations and experiment (Fig. 5.3D). The root mean
square inter-residue distance between simulated positions 172 and 245 is 59.1 Å, which is within the
experimental error of the single-molecule observations. Normalized distance map shows a number of
regions of repulsion, notably that the RBD repels the N-terminal part of the LINK and the
dimerization domain repels the C-terminal part of the LINK (Fig. 5.3E). We tentatively suggest this
may reflect sequence properties chosen to prevent aberrant interactions between the LINK and the two
folded domains. In the LINK-only simulations we identified two regions that form transient helices
at low populations (20-25%), although these are much less prominent in the context of
the full length protein (Fig. 5.3F). Those helices encompass a serine-arginine (SR) rich
region known to mediate both protein-protein and protein-RNA interaction, and leads to the
alignment of three arginine residues along one face of a helix. The second helix (H4) is a
leucine/alanine-rich hydrophobic helix which may contribute to oligomerization, or act as a helical
recognition motif for other protein interactions (notably as a nuclear export signal for Crm1, see
Discussion).
5.3.3 The CTD engages in transient but non-negligible interactions with the dimerization
domain.
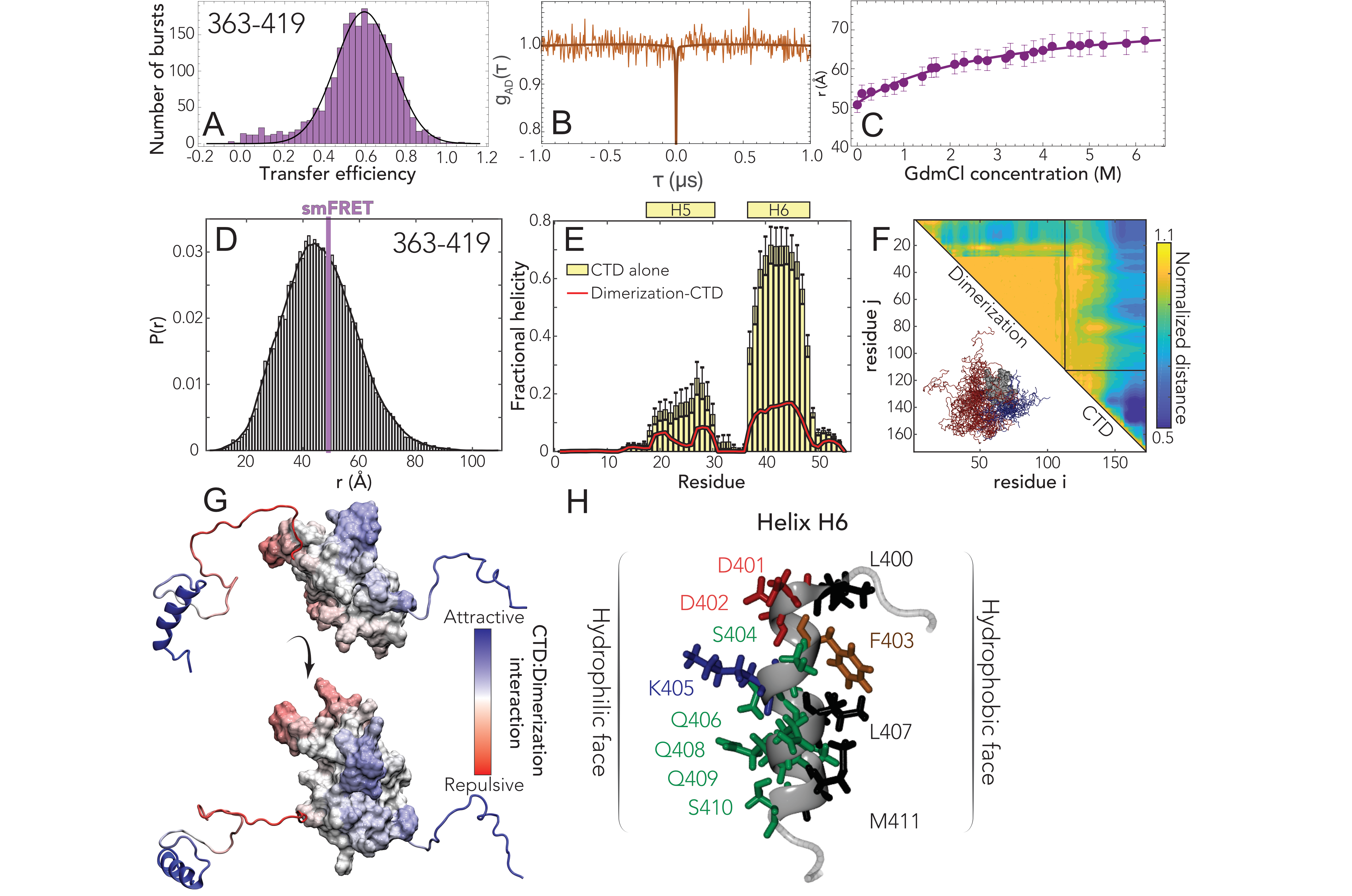
Finally, we turned to the CTD. Single-molecule FRET experiments again reveal a single population with a mean transfer
efficiency of 0.59
0.03 (Fig. 5.4A) and the denaturant dependence follows the expected trend for a disordered region,
with a shift of the transfer efficiency toward lower values (Fig. D.6,D.8), from 0.59 to
0.35. Interestingly, when studying the denaturant dependence of the protein, we noticed
that the width of the distribution increases while moving toward native conditions. This
suggests that the protein may form transient contacts or adopt local structure. To investigate
this aspect, we turned to the investigation of the dynamics. Though the comparison of
the fluorophore lifetimes against transfer efficiency (Fig. D.7c) appears to support a
dynamic nature underlying this population, nanosecond FCS reveals a flat acceptor-donor
cross-correlation on the ns timescale (Fig. 5.4B). However, inspection of the donor-donor and
acceptor-acceptor autocorrelations reveal a correlated decay with a characteristic time of 240
50 ns.
This is different from that expected for a completely static system such as polyprolines [330], where
the donor-donor and acceptor-acceptor autocorrelation are also flat. An increase in the autocorrelations
can be observed for static quenching of the dyes with aromatic residues.Interestingly, donor dye
quenching can also contribute to a positive amplitude in the donor-acceptor correlation [331, 332].
Therefore, a plausible interpretation of the flat cross-correlation data is that we are observing two
populations in equilibrium whose correlations (one anticorrelated, reflecting conformational
dynamics, and one correlated, reflecting quenching due contact formation) compensate each
other.
To further investigate the possible coexistence of these different species, we performed
ns-FCS at 0.2 M GdmCl, where the width of the FRET population starts decreasing and
the mean transfer efficiency is slightly shifted to larger values, under the assumption
that the decreased width of the population reflects reduced interactions. Indeed, the
cross-correlation of ns-FCS reveals a dynamic behavior with a reconfiguration time
= 70
15 ns (Fig.
D.11). Based on these observations, we suggest that a very similar disordered population to the one
observed at 0.2 M is also present under native conditions, but in equilibrium with a quenched species
that forms long-lived contacts. Under the assumption that the mean transfer efficiency still originates
(at least partially) from a dynamic distribution, the estimate of the inter-residue root-mean-square distance
is = 51
2 Å
(= 6.1
0.4 Å) for a Gaussian
chain distribution and
= 49 2
Å (=
5.6 0.4
Å) for the SAW model (see SI). However, some caution should be used when interpreting these
numbers since we know there is some contribution from fluorophore quenching, which may in turn
contribute to an underestimate of the effective transfer efficiency [333].
We again obtained good agreement between all-atom Monte Carlo simulations and experiment
(Fig. 5.4D). We identified two transient helices, one (H5) is minimally populated but the
second (H6) is more highly populated in the IDR-only simulation and still present at
20% in
the folded state simulations (Fig. 5.4E). The difference reflects the fact that several of the
helix-forming residues interact with the dimerization domain, leading to a competition between helix
formation and intramolecular interaction. Scaling maps reveal extensive intramolecular interaction by
the residues that make up H6, both in terms of local intra-IDR interactions and interaction with the
dimerization domain (Fig. 5.4F). Mapping normalized distances onto the folded structure
reveals that interactions occur primarily with the N-terminal portion of the dimerization
domain (Fig. 5.4G). As with the LINK and the NTD, a positively charged set of residues
immediately adjacent to the folded domain in the CTD drive repulsion between this region and the
dimerization domain. H6 is the most robust helix observed across all three IDRs, and is
a perfect amphipathic helix with a hydrophobic surface on one side and charged/polar
residues on the other (Fig. 5.4H). The cluster of hydrophobic residues in H6 engage in
intramolecular contacts and offer a likely physical explanation for the complex lifetime
data.
5.3.4 N protein undergoes phase separation with RNA.
Over the last decade, biomolecular condensates formed through phase separation have
emerged as a new mode of cellular organization [334, 335, 336, 337]. Given the high
interaction valency and the presence of molecular features similar to other proteins we had
previously studied, we anticipated that N protein would undergo phase separation with
RNA [338, 339, 340].
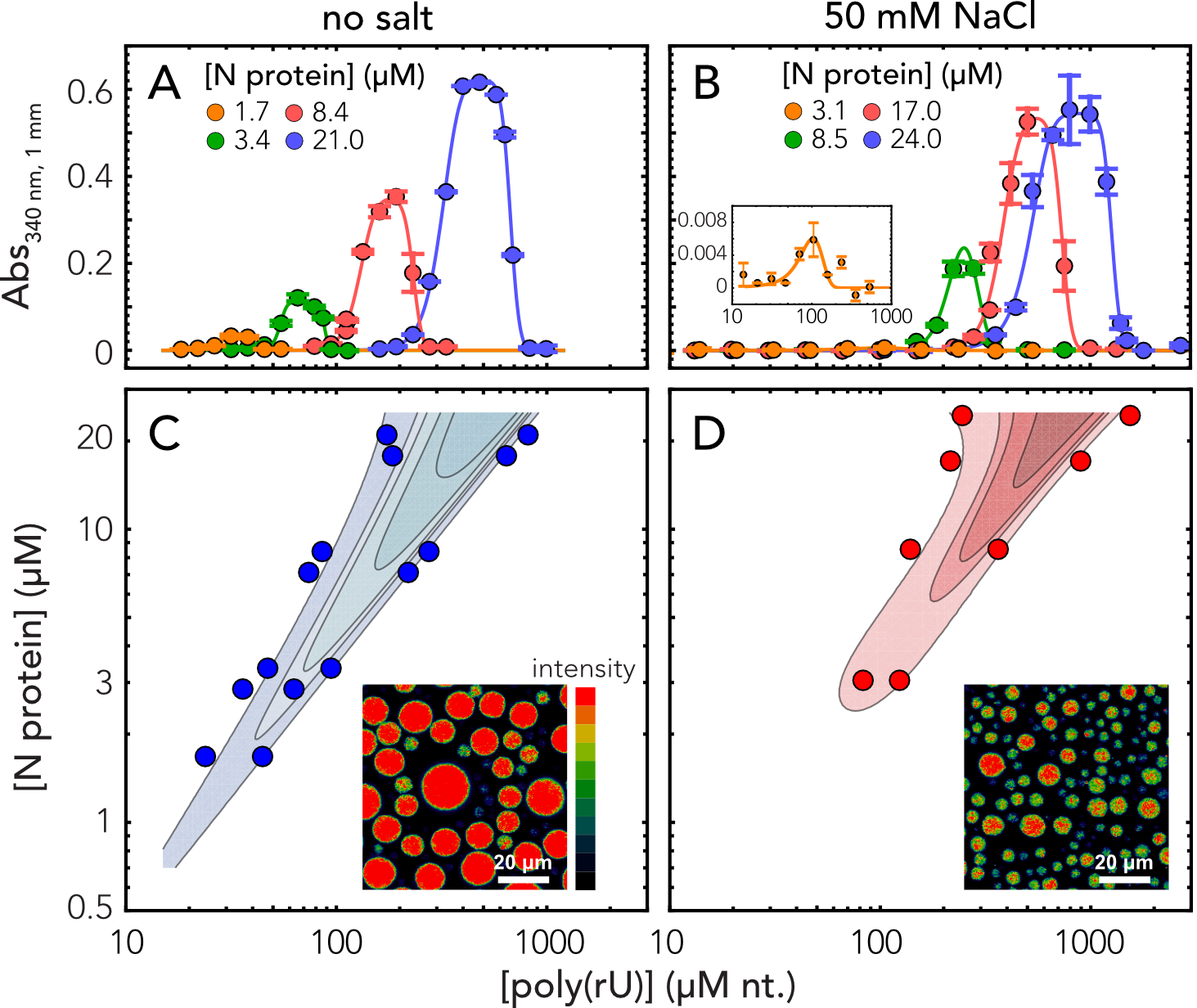
In line with this expectation, we observed robust droplet formation with homopolymeric RNA (Fig.
5.5A-B) under native buffer conditions (50 mM Tris) and at higher salt concentration (50 mM NaCl).
Turbidity assays at different concentrations of protein and poly(rU) (200-250 nucleotides) demonstrate
the classical reentrant phase behavior expected for a system undergoing heterotypic interaction (Fig.
5.5C-D). It is to be noted that turbidity experiments do not exhaustively cover all the conditions for
phase separation and are only indicative of the low-boundary concentration regime explored in the
current experiments. In particular, turbidity experiments do not provide a measurement of tie-lines,
though they are inherently a reflection of the free energy and chemical potential of the solution
mixture [341]. Interestingly, phase separation occurs at relatively low concentrations, in the low
M
range, which are compatible with physiological concentration of the protein and nucleic acids.
Though increasing salt concentration results in an upshift of the phase boundaries, one has
to consider that in a cellular environment this effect might be counteracted by cellular
crowding.
One peculiar characteristic of our measured phase-diagram is the narrow regime of conditions in
which we observe phase separation of nonspecific RNA at a fixed concentration of protein. This leads
us to hypothesize that the protein may have evolved to maintain a tight control of concentrations at
which phase separation can (or cannot) occur. Interestingly, when rescaling the turbidity curves as a
ratio between protein and RNA, we find all the curve maxima aligning at a similar stoichiometry,
approximately 20 nucleotides per protein in absence of added salt and 30 nucleotides when adding 50
mM NaCl (Fig. D.12). These ratios are in line with the charge neutralization criterion proposed
by Banerjee et al., since the estimated net charge of the protein at pH 7.4 is +24 [342].
Finally, given we observed phase separation with poly(rU), the behavior we are observing is
likely driven by relatively nonspecific protein:RNA interactions. In agreement, work from
the Gladfelter [298], Fawzi [299], Zweckstetter [300], and Yildiz (unpublished) labs
have also established this phenomenon across a range of solution conditions and RNA
types.
Having established phase separation through a number of assays, we wondered what -if any-
physiological relevance this may have for the normal biology of SARS-CoV-2.
5.3.5 A simple polymer model shows symmetry-breaking can facilitate multiple metastable
single-polymer condensates instead of a single multi-polymer condensate.
Why might phase separation of N protein with RNA be advantageous to SARS-CoV-2? One possible
model is that large, micron-sized cytoplasmic condensates of N protein with RNA form
through phase separation and play a role in genome packaging. These condensates may act as
molecular factories that help concentrate the components for pre-capsid assembly (where we
define a pre-capsid here simply as a species that contains a single copy of the genome
with multiple copies of the associated N protein), a model that has been proposed in other
viruses [343].
However, given that phase separation is unavoidable when high concentrations of multivalent species
are combined, we propose that an alternative interpretation of our data is that in this context, phase
separation is simply an inevitable epiphenomenon that reflects the inherent multi-valency of the N
protein for itself and for RNA. This poses questions about the origin of specificity for viral genomic
RNA (gRNA), and, of focus in our study, how phase separation might relate to a single genome
packaging through RNA compaction.
Given the expectation of a single genome per virion, we reasoned SARS-CoV-2 may have evolved a
mechanism to limit phase separation with gRNA (i.e. to avoid multi-genome condensates), with a
preference instead for single-genome packaging (single-genome condensates). This mechanism may
exist in competition with the intrinsic phase separation of the N protein with other nonspecific RNAs
(nsRNA).
One possible way to limit phase separation between two components (e.g. gRNA/nsRNA and
N protein) is to ensure the levels of these components are held at a sufficiently low total
concentration such that the phase boundary is never crossed. While possible, suc,h a regulatory
mechanism is at the mercy of extrinsic factors that may substantially modulate the saturation
concentration [?, 344, 345, 346]. Furthermore, not only must phase separation be prevented,
but gRNA compaction should also be promoted through the binding of N protein. In this
scenario, the affinity between gRNA and N protein plays a central role in determining the
required concentration for condensation of the macromolecule (gRNA) by the ligand (N
protein).
Given a defined valence of the system components, phase boundaries are encoded by the strength of
interaction between the interacting domains in the components. Considering a long polymer (e.g.
gRNA) with proteins adsorbed onto that polymer as adhesive points (“stickers”), the physics of
associative polymers predicts that the same interactions that cause phase separation will also control
the condensation of individual long polymers [338, 347, 348, 349, 350, 351]. With this in mind,
we hypothesized that phase separation is reporting on the physical interactions that underlie genome
compaction.
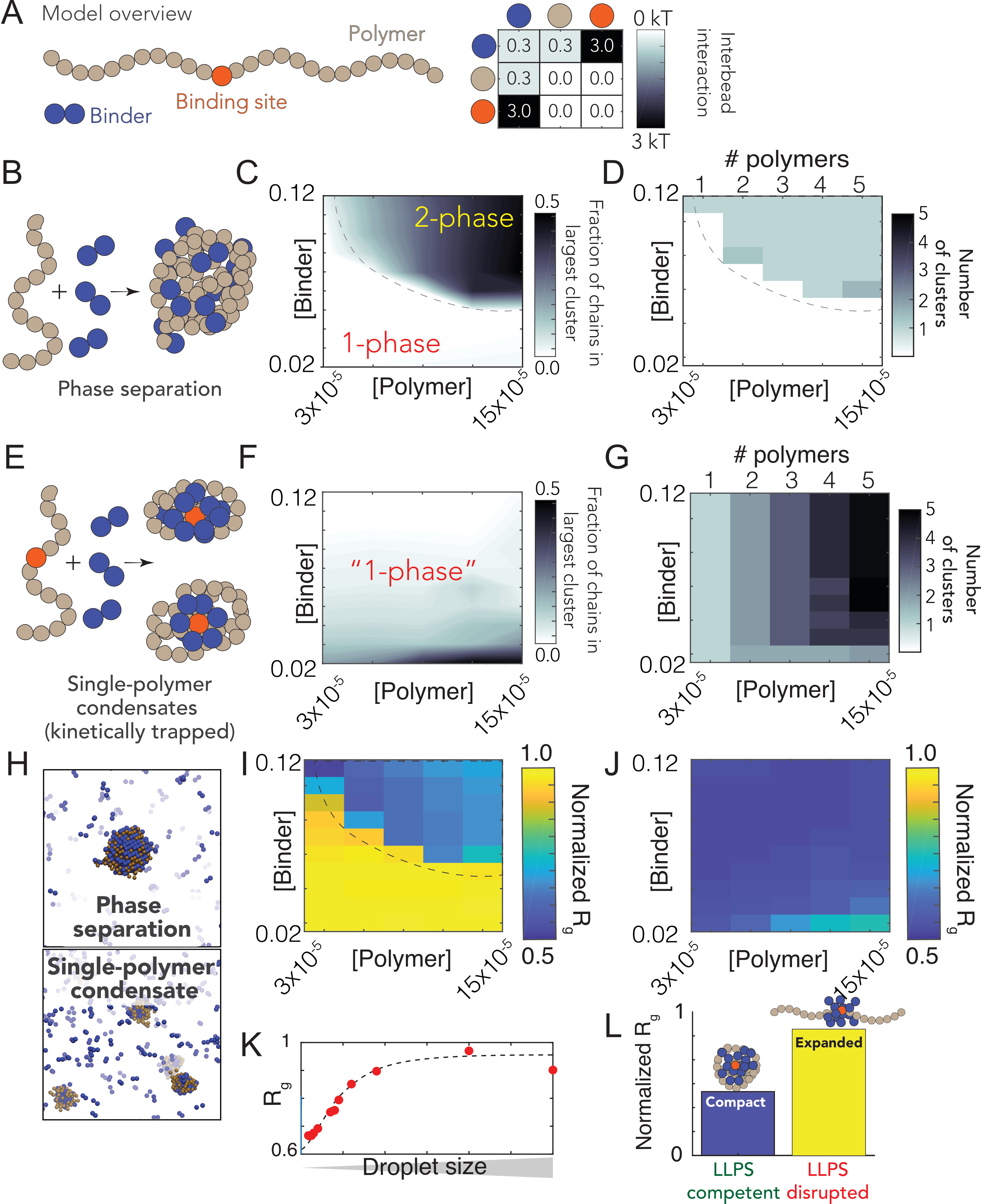
To explore this hypothesis, we developed a simple computational model where the interplay between
compaction and phase separation could be explored. Our setup consists of two types of species: long
multivalent polymers and short multivalent binders (Fig. 5.6A). All interactions are isotropic and each
bead is inherently multivalent as a result. In the simplest instantiation of this model, favourable
polymer:binder and binder:binder interactions are encoded, mimicking the scenario in which a binder
(e.g. a protein) can engage in nonspecific polymer (RNA) interaction as well as binder-binder
(protein-protein) interaction.
Simulations of binder and polymer undergo phase separation in a concentration-dependent manner,
as expected (Fig. 5.6B,C). Phase separation gives rise to a single large spherical cluster
with multiple polymers and binders (Fig. 5.6D, 5.6H). For a homopolymer, the balance of
chain-compaction and phase separation is determined in part through chain length and binder
. In our system
the polymer is largely unbound in the one-phase regime (suggesting the concentration of ligand in the one-phase
space is below the )
but entirely coated in the two-phase regime, consistent with highly-cooperative binding behavior. In
the limit of long, multivalent polymers with multivalent binders, the sharpness of the coil-to-globule
transition is such that an effective two-state description of the chain emerges, in which the chain is
either expanded (non-phase separation-competent) OR compact (coated with binders, phase separation
competent).
In light of these observations, we wondered if a break in the symmetry between intra- and
inter-molecular interactions would be enough to promote single-polymer condensation in
the same concentration regime over which we had previously observed phase separation.
Symmetry breaking in our model is achieved through a single high-affinity binding site (Fig.
5.6A). We choose this particular mode of symmetry-breaking to mimic the presence of a
packaging signal -a region of the genome that is essential for efficient viral packaging-
an established feature in many viruses (including coronaviruses) although we emphasize
this is a general model, as opposed to trying to directly model gRNA with a packaging
signal [352, 353, 354].
We performed identical simulations to those in Fig. 5.6C-D using the same system with polymers that
now possess a single high affinity binding site (Fig. 5.6E). Under these conditions we did not observe
large phase separated droplets (Fig. 5.6F). Instead, each individual polymer undergoes collapse to
form a single-polymer condensate (Fig. 5.6E). Collapse is driven by the recruitment of binders to the
high-affinity site, where they “coat” the chain, forming a local cluster of binders on the polymer. This
cluster is then able to interact with the remaining regions of the polymer through weak
“nonspecific” interactions, the same interactions that drove phase separation in Fig. 5.6B,C,D.
Symmetry breaking is achieved because the local concentration of binder around the site is
high, such that intramolecular interactions are favoured over intermolecular interaction.
This high local concentration also drives compaction at low binder concentrations. As a
result, instead of a single multi-polymer condensate, we observe multiple single-polymers
condensates, where the absolute number matches the number of polymers in the system (Fig.
5.6G).
Our results can also be cast in terms of two distinct concentration (phase) boundaries - one for binder:high affinity
site interaction (),
and a second boundary for “nonspecific” binder:polymer interactions
() at a higher
concentration.
reflects the boundary observed in Fig. 5.6C that delineated the one and two-phase regimes. At global concentrations
below , (but
above )
the clustering of binders at a high affinity site raises the apparent local concentration of binders above
, from
the perspective of other beads on the chain. In this way, a local high affinity binding site can drive
“local” phase separation of a single polymer.
The high affinity binding site polarizes the single-polymer condensate, such that they are organized,
recalcitrant to fusion, and kinetically stable. A convenient physical analogy is that of a micelle, which
are non-stoichiometric stable assemblies. Even for micelles that are far from their optimal size, fusion
is slow because it requires substantial molecular reorganization and the breaking of stable
interactions [355, 356].
Finally, we ran simulations under conditions in which binder:polymer interactions were reduced,
mimicking the scenario in which non-specific protein:RNA interactions are inhibited (Fig. 5.6L).
Under these conditions no phase separation occurs for polymers that lack a high-affinity binding site,
while for polymers with a high-affinity binding site no chain compaction occurs (in contrast to when
binder:polymer interactions are present, see Fig. 5.6J). This result illustrates how phase separation
offers a convenient readout for molecular interactions that might otherwise be challenging to
measure.
We emphasize that our conclusions from simulations are subject to the parameters in our model. We
present these results to demonstrate an example of “how this single-genome packaging could be
achieved”, as opposed to the much stronger statement of proposing “this is how it is” achieved. Recent
elegant work by Ranganathan and Shakhnovich identified kinetically arrested microclusters, where
slow kinetics result from the saturation of stickers within those clusters [357]. This is completely
analogous to our results (albeit with homotypic interactions, rather than heterotypic interactions),
giving us confidence that the physical principles uncovered are robust and, we tentatively
suggest, quite general. Future simulations are required to systematically explore the details of
the relevant parameter space in our system. However, regardless of those parameters, our
model does establish that if weak multivalent interactions underlie the formation of large
multi-polymer droplets, those same interactions cannot also drive polymer compaction inside the
droplet
5.4 Discussion
The nucleocapsid (N) protein from SARS-CoV-2 is a multivalent RNA binding protein critical for
viral replication and genome packaging [289, 291]. To better understand how the various folded and
disordered domains interact with one another, we applied single-molecule spectroscopy and all-atom
simulations to perform a detailed biophysical dissection of the protein, uncovering several putative
interaction motifs. Furthermore, based on both sequence analysis and our single-molecule
experiments, we anticipated that N protein would undergo phase separation with RNA. In agreement
with this prediction, and in line with work from the Gladfelter and Yildiz groups working
independently from us, we find that N protein robustly undergoes phase separation in vitro
with model RNA under a range of different salt conditions. Using simple polymer models,
we propose that the same interactions that drive phase separation may also drive genome
packaging into a dynamic, single-genome condensate. The formation of single-genome
condensates (as opposed to multi-genome droplets) is influenced by the presence of one (or more)
symmetry-breaking interaction sites, which we tentatively suggest could reflect packaging signals in
viral genomes.
5.4.1 All three IDRs are highly dynamic
Our single-molecule experiments and all-atom simulations are in good agreement with one another,
and reveal that all three IDRs are extended and highly dynamic. Simulations suggest the NTD may
interact transiently with the RBD, which offers an explanation for the slightly slowed reconfiguration
time measured by nanosecond FCS. The LINK shows rapid rearrangement, demonstrating the RBD
and dimerization domain are not interacting. Finally, we see more pronounced interaction
between the CTD and the dimerization domain, although these interactions are still highly
transient.
Single-molecule experiments and all-atom simulations were performed on monomeric versions of the
protein, yet N protein has previously been shown to undergo dimerization and form higher-order
oligomers in the absence of RNA [310]. To assess the formation of oligomeric species, we use a
combination of nativePAGE, crosslinking and FCS experiments (see Fig. D.13). These experiments
also verified that under the conditions used for single-molecule experiments the protein exists only as
a monomer.
5.4.2 Simulations identify multiple transient helices
We identified a number of transient helical motifs which provide structural insight into
previously characterized molecular interactions. Transient helices are ubiquitous in viral
disordered regions and have been shown to underlie molecular interactions in a range of
systems [343, 358, 359, 360].
Transient helix H2 (in the NTD) and H3 (in the LINK) flank the RBD and organize a set of arginine
residues to face the same direction (Fig. 5.2E). Both the NTD and LINK have been shown to drive
RNA binding, such that we propose these helical arginine-rich motifs (ARMs) may engage in both
both nonspecific binding and may also contribute to RNA specificity, as has been proposed
previously [303, 361, 362]. The serine-arginine SR-region (which includes H3) has been previously
identified as engaging in interaction with a structured acidic helix in Nsp3 in the model coronavirus
MHV, consistent with an electrostatic helical interaction [363, 364]. Recent NMR data also shows
excellent agreement with our results, identifying a transient helix that shows 1:1 overlap with
H3 [300].The SR-region is necessary for recruitment to replication-transcription centers (RTCs) in
MHV, and also undergoes phosphorylation, setting the stage for a complex regulatory system awaiting
exploration [365, 366].
Transient helix H4 (Fig. 5.3H), was previously predicted bioinformatically and identified as a
conserved feature across different coronaviruses [303]. Furthermore, the equivalent region was
identified in SARS coronavirus as a nuclear export signal (NES), such that we suspect this too is a
classical Crm1-binding leucine-rich NES [367].
Transient helix H6 is an amphipathic helix with a highly hydrophobic face (Fig. 5.4H). Recent
hydrogen-deuterium exchange mass spectrometry also identified H6 [315]. Residues in this region
have previously been identified as mediating M-protein binding in other coronaviruses, such that we
propose H6 underlies that interaction [368, 369, 370]. Recent work has also identified amphipathic
transient helices in disordered proteins as interacting directly with membranes, such that an additional
(albeit entirely speculative) role could involve direct membrane interaction, as has been observed in
other viral phosphoproteins [371, 372].
5.4.3 The physiological relevance of nucleocapsid protein phase separation in SARS-CoV-2
physiology
Our work has revealed that SARS-CoV-2 N protein undergoes phase separation with RNA when
reconstituted in vitro. The solution environment and types of RNA used in our experiments are very
different from the cytoplasm and viral RNA. However, similar results have been obtained in published
and unpublished work by several other groups under a variety of conditions, including via in cell
experiments (Yildiz group, unpublished) [298, 299, 300]. Taken together, these results demonstrate
that N protein can undergo bona fide phase separation, and that N protein condensates can form in
cells. Nevertheless, the complexity introduced by multidimensional linkage effects in vivo could
substantially influence the phase behavior and composition of condensates observed in the
cell [346, 350, 373]. Of note, the regime we have identified in which phase separation occurs (Fig.
5.5) is remarkably relatively narrow, a prerequisite for the assembly of virion particles containing a
single viral genome.
Does phase separation play a physiological role in SARS-CoV-2 biology? Phase separation has been
invoked or suggested in many different viral contexts to date [374, 375, 376, 377, 378]. In
SARS-CoV-2, one possible model suggests phase separation may drive recruitment of components to
viral replication sites, although how this dovetails with the fact that replication occurs in
double-membrane bound vesicles (DMVs) remains to be explored [300, 379]. An alternative (and
non-mutually exclusive) model is one in which phase separation catalyzes nucleocapsid
polymerization, as has been proposed in elegant work on measles virus [343]. Here, the process
of phase separation is decoupled from genome packaging, where gRNA condensation
occurs through association with a helical nucleocapsid. If applied to SARS-CoV-2, such a
model would suggest that (1) initially N protein and RNA phase separate in the cytosol, (2)
some discrete pre-capsid state forms within condensates and, (3) upon maturation, the
pre-capsid is released from the condensate and undergoes subsequent virion assembly
by interacting with the membrane-bound M, E, and S structural proteins at the ER-Golgi
intermediate compartment (ERGIC). While this model is attractive it places a number of
constraints on the physical properties of this pre-capsid, not least that the ability to escape the
“parent” condensate dictates that the assembled pre-capsid must interact less strongly with the
condensate components than in the unassembled state. This requirement introduces some
thermodynamic complexities: how is a pre-capsid state driven to assemble if it is necessarily
less stable than the unassembled pre-capsid, and how is incomplete or abortive pre-capsid
formation avoided if – as assembly occurs – the pre-capsid becomes progressively less
stable?
A phase separation and assembly model raises additional questions, such as the origins of specificity
for recruitment of viral proteins and viral RNA, the kinetics of pre-capsid-assembly within a large
condensate, and preferential packaging of gRNA over sub-genomic RNA. None of these questions are
unanswerable, nor do they invalidate this model, but they should be addressed if the physiological
relevance of large cytoplasmic condensates is to be further explored in the context of virion
assembly.
Our preferred interpretation is that N protein has evolved to drive genome compaction for
packaging (Fig. 5.7). In this model, a single-genome condensate forms through N protein gRNA
interaction, driven by a small number of high-affinity sites. This (meta)-stable single-genome
condensate undergoes subsequent maturation, leading to virion assembly. In this model,
condensate-associated N proteins are in exchange with a bulk pool of soluble N protein, such that the
interactions that drive compaction are heterogeneous and dynamic. Our model provides a
physical mechanism in good empirical agreement with data for N protein oligomerization and
assembly [380, 381, 382]. Furthermore, the resulting condensate is then in effect a multivalent
binder for M protein, which interacts with N directly, and may drive membrane curvature and budding
in a manner similar to that proposed by Bergeron-Sandoval and Michnick (though with a different
directionality of the force) and in line with recent observations from cryo electron tomography
(cryoET) [379, 383, 384, 385].
An open question pertains to specificity of packaging gRNA while excluding other RNAs. One
possibility is for two high-affinity N-protein binding sites to flank the 5’ and 3’ ends of the genome,
whereby only RNA molecules with both sites are competent for compaction A recent map of N
protein binding to gRNA has revealed high-affinity binding regions at the 5’ and 3’ ends of the
gRNA, in good agreement with this qualitative prediction [298]. Alternatively only gRNA
condensates may possess the requisite valency to drive virion budding through interaction
with M at the cytoplasmic side of the ERGIC, offering a physical selection mechanism for
budding.
Genome compaction through dynamic multivalent interactions would be especially relevant for
coronaviruses, which have extremely large single-stranded RNA genomes. This is evolutionarily
appealing, in that as the genome grows larger, compaction becomes increasingly efficient, as
the effective valence of the genome is increased [348, 349]. The ability of multivalent
disordered proteins to drive RNA compaction has been observed previously in various
contexts [292, 386]. Furthermore, genome compaction by RNA binding protein has been
proposed and observed in other viruses [382, 387, 388], and the SARS coronavirus N
protein has previously been shown to act as an RNA chaperone, an expected consequence of
compaction to a dynamic single-RNA condensate that accommodates multiple N proteins with a
single RNA [292, 389]. Furthermore, previous work exploring the ultrastructure of phase
separated condensates of G3BP1 and RNA through simulations and cryoET revealed a
beads-on-a-string type architecture, mirroring recent results for obtained from cryoET of
SARS-CoV-2 virions [339, 379].
N protein has been shown to interact directly with a number of proteins studied in the context of
biological phase separation which may influence assembly in vivo [283, 298, 338, 345, 390]. In
particular, G3BP1-an essential stress-granule protein that undergoes phase separation-was recently
shown to co-localize with overexpressed N protein [300, 339, 345, 391]. G3BP1 interaction may be
part of the innate immune response, leading to stress-granule formation, or alternatively N protein may
attenuates the stress response by sequestering G3BP1, depleting the cytosolic pool, and preventing
stress granule formation, as has been shown for HIV-1 [378].
Our model is also in good empirical agreement with recent observations made for other viruses [392].
Taken together, we speculate that viral packaging may -in general- involve an initial genome
compaction through multivalent protein:RNA and protein:protein interactions, followed by a
liquid-to-solid transition in cases where well-defined crystalline capsid structures emerge.
Liquid-to-solid transitions are well established in the context of neurodegeneration with respect to
disease progression [393, 394, 395]. Here we suggest nature is leveraging those same principles as
an evolved mechanism for monodisperse particle assembly.
Regardless of if phase separated condensates form inside cells, all available evidence suggests phase
separation is reporting on a physiologically important interaction that underlies genome compaction
(Fig. 5.6L). With this in mind, from a biotechnology standpoint, phase separation may be a convenient
readout for in vitro assays to interrogate protein:RNA interaction. Regardless of which
model is correct, N protein:RNA interaction is key for viral replication. As such, phase
separation provides a macroscopic reporter on a nanoscopic phenomenon, in line with previous
work [338, 348, 396, 397]. In this sense, we believe the therapeutic implications of understanding
and modulating phase separation here (and elsewhere in biology) are conveniently decoupled
from the physiological relevance of actual, large phase separated “liquid droplets”, but
instead offer a window into the underlying physical interactions that lead to condensate
formation.
5.4.4 The physics of single polymer condensates
Depending on the molecular details, single-polymer condensates may be kinetically stable (but
thermodynamically unstable, as in our model simulations) or thermodynamically stable. Delineation
between these two scenarios will depend on the nature, strength, valency and anisotropy of the
interactions. It is worth noting that from the perspective of functional biology, kinetic stability may be
essentially indistinguishable from thermodynamic stability, depending on the lifetime of a metastable
species.
It is also important to emphasize that at higher concentrations of N protein and/or after a
sufficiently long time period we expect robust phase separation with viral RNA, regardless
of the presence of a symmetry-breaking site. Symmetry breaking is achieved when the
apparent local concentration of N protein (from the “perspective” of gRNA) is substantially
higher than the actual global concentration. As effective local and global concentrations
approach one another, the entropic cost of intra-molecular interaction is outweighed by the
availability of inter-molecular partners. On a practical note, if the readout in question is
the presence/absence of liquid droplets, a high-affinity site may be observed as a shift in
the saturation concentration which, confusingly, could either suppress or enhance phase
separation. Further, if single-genome condensates are kinetically stable and driven through
electrostatic interactions, we would expect a complex temperature dependence, in which
larger droplets are observed at higher temperature (up to some threshold). Recent work is
showing a strong temperature-dependence of phase separation is consistent with these
predictions [298].
Finally, we note no reason to assume single-RNA condensates should be exclusively the purview of
viruses. RNAs in eukaryotic cells may also be processed in these types of assemblies, as opposed to in
large multi-RNA RNPs. The role of RNA:RNA interactions both here and in other systems is also of
particular interest and not an aspect explored in our current work, but we anticipate may play a key
role in the relevant biology.
5.5 Methods
5.5.1 All atom simulations
All-atom Monte Carlo simulations were performed with the ABSINTH implicit solvent model and
CAMPARI simulation engine (http://campari.sourceforge.net/) [398, 399] with the solution ion
parameters of Mao et al. [400]. Simulations were performed using movesets and Hamiltonian
parameters as reported previously [338, 401]. All simulations were performed in sufficiently large
box sizes to prevent finite size effects (where box size varies from system to system). For
simulations with IDRs in isolation all degrees of freedom available in CAMPARI are sampled.
For simulations with folded domains with IDRs, the backbone dihedral angles in folded
domains are not sampled, such that folded domains remain structurally fixed (although
sidechains are fully sampled). The IDR has backbone and sidechain degrees of freedom
sampled.
All-atom molecular dynamics simulations were performed using GROMACS, using the FAST
algorithm in conjunction with the Folding@home platform [37, 222, 35]. Post-simulation analysis
was performed with Enspara [39]. For additional simulation details see the supplementary
information.
5.5.2 Coarse-grained Polymer Simulations
Coarse-grained Monte Carlo simulations were performed using the PIMMS simulation engine [402].
All simulations were performed in a 70 x 70 x 70 lattice-site box. The results averaged over the final
20% of the simulation to give average values at equivalent states. The “polymer” is represented as a
61-residue polymer with either a central high-affinity binding site or not. The binder is a 2-bead
species. Every simulation was run for 20 x 109 Monte Carlo steps, with four independent replicas.
Bead interaction strengths were defined as shown in Fig. 5.6A. For additional simulation details see
the supplementary information.
5.5.3 Protein Expression, purification, and labeling.
SARS-CoV-2 Nucleocapsid protein (NCBI Reference Sequence: YP_009724397.2) including an N
term extension containing His9-HRV 3C protease site was cloned into the BamHI EcoRI sites in the
MCS of pGEX-6P-1 vector (GE Healthcare). Site-directed mutagenesis was performed on the
His9-SARS-CoV-2 Nucleocapsid pGEX vector to create M1C R68C, Y172C T245C, and F363C
A419C variant N protein constructs and sequences were verified using Sanger sequencing. All variants
were expressed recombinantly in BL21 Codon-plus pRIL cells (Agilent) or Gold BL21(DE3) cells
(Agilent) and purified using a FF HisTrap column. The GST-His9-N tag was then cleaved using HRV
3C protease and further purified to remove the cleaved tag. Finally, purified N protein variants were
analyzed using SDS-PAGE and verified by electrospray ionization mass spectrometry (LC-MS).
Activity of the protein was assessed by testing whether the protein is able to bind and condense
nucleic acids (see phase-separation experiments) as well as to form dimers (see oligomerization in
SI).
All Nucleocapsid variants were labeled with Alexa Fluor 488 maleimide and Alexa Fluor 594
maleimide (Molecular Probes) under denaturing conditions following a two-step sequential labeling
procedure (see SI).
5.5.4 Single-molecule fluorescence spectroscopy.
Single-molecule fluorescence measurements were performed with a Picoquant MT200 instrument
(Picoquant, Germany). FRET experiments were performed by exciting the donor dye with a laser power of
100 W
(measured at the back aperture of the objective). For pulsed interleaved excitation of donor and
acceptor, the power used for exciting the acceptor dye was adjusted to match the acceptor emission
intensity to that of the donor (between 50 and 70 mW). Single-molecule FRET efficiency histograms
were acquired from samples with protein concentrations between 50 pM and 100 pM and the
population with stoichiometry corresponding to 1:1 donor:acceptor labeling was selected. Trigger
times for excitation pulses (repetition rate 20 MHz) and photon detection events were stored with 16
ps resolution. For FRET-FCS, samples of double-labeled protein with a concentration of 100 pM
were excited by either the diode laser or the supercontinuum laser at the powers indicated
above.
All samples were prepared in 50 mM Tris pH 7.32, 143 mM
-mercaptoethanol
(for photoprotection), 0.001% Tween 20 (for limiting surface adhesion) and GdmCl at the reported
concentrations. All measurements were performed in uncoated polymer coverslip cuvettes (Ibidi,
Wisconsin, USA), which significantly decrease the fraction of protein adhering to the surface
(compared to normal glass cuvettes) under native conditions. For comparison, experiments have been
performed also in glass cuvette coated with PEG, which provided analogous results to the
polymeric cuvette. Each sample was measured for at least 30 min at room temperature (295
0.5 K)
(see appendix E).
5.6 Acknowledgements
We thank Amy Gladfelter, Christiane Iserman, Christine Roden, Ahmet Yildiz, Amanda Jack,
Luke Ferro, Steve Michnick, Pascale Legault, and Jim Omichinski for sharing data and
extensive discussion. We also thank Rohit Pappu for placing our groups in contact with one
another.
We thank the labs of John Cooper, Carl Frieden, and Silvia Jansen for providing some of the
reagents we have used in this work. We thank Ben Schuler and Daniel Nettels for developing,
maintaining, and sharing with us the software package used to analyze the single-molecule
data.
J.C. and J.J.A are supported by NIGMS R25 IMSD Training Grant GM103757. We are grateful to the
citizen-scientists of Folding@home for donating their computing resources. G.R.B holds an NSF
CAREER Award MCB-1552471, NIH R01GM12400701, a Career Award at the Scientific Interface
from the Burroughs Wellcome Fund, and a Packard Fellowship for Science and Engineering from The
David and Lucile Packard Foundation. []
A.S.H. is a scientific consultant with Dewpoint Therapeutics.