Chapter 4
Discovery of a cryptic allosteric site in Ebola’s ‘undruggable’ VP35 protein using
simulations and experiments
This chapter is adapted from the following publication:
Cruz, M.A.and
Frederick, T.E.,
Singh, S., Vithani, N., Zimmerman, M.I., Porter, J.R., Moeder, K.E., Amarasinghe, G.K., and Bowman,
G.R., Discovery of a cryptic allosteric site in Ebola’s ’undruggable’ VP35 protein using simulations
and experiments. Preprint on BioRxiv https://doi.org/10.1101/2020.02.09.940510 [54]
Authors
contributed equally to this work
In this work, my work analyzing the allostery in VP35 and the coupling between the cryptic pocket
opening and binding interface is presented in figure 4.3 and appendix figure C.2.
4.1 Abstract
Many proteins are classified as ‘undruggable,’ especially those that engage in protein-protein and
protein-nucleic acid interactions. Discovering ‘cryptic’ pockets that are absent in available structures
but open due to protein dynamics could provide new druggable sites. Here, we integrate simulations
and experiments to search for cryptic pockets in Ebola viral protein 35 (VP35). VP35 plays
essential roles in Ebola’s replication cycle, including binding the viral RNA genome to block a
host’s innate immunity. However, VP35 has so far proved undruggable. Using adaptive
sampling simulations and allosteric network detection algorithms, we uncover a cryptic pocket
that is allosterically coupled to VP35’s key RNA-binding interface. Experimental tests
corroborate the predicted pocket and confirm that stabilizing the open form allosterically
disrupts RNA binding. These results demonstrate simulations’ power to characterize hidden
conformations and dynamics, uncovering cryptic pockets and allostery that present new therapeutic
opportunities.
4.2 Introduction
Many proteins have proved so difficult to target with small molecule drugs that they are often
classified as undruggable, greatly limiting the scope of drug design efforts. In fact, up to 85% of
human proteins have been classified as undruggable because their folds are thought to lack binding
pockets where small molecules can bind with the affinity and specificity required for drug
design [239]. Many undruggable proteins predominantly participate in protein-protein interactions
(PPIs) and protein-nucleic acid interactions (PNIs) [72, 240]. In contrast to the binding pockets that
many enzymes and receptors use to bind their small molecule ligands, the large flat interfaces involved
in PPIs and PNIs do not lend themselves to forming many favorable interactions with small drug-like
molecules. As a result, PPIs and PNIs are often considered intractable targets even when
there is strong evidence that disrupting these interactions would be of great therapeutic
value.
Cryptic pockets could provide new opportunities to target undruggable proteins [41, 241], but
realizing this potential remains challenging. Such pockets are absent in available experimental
structures because they only form in a subset of excited states that arise due to protein dynamics.
Cryptic sites can serve as valuable drug targets if they coincide with key functional sites, as can
cryptic allosteric sites that are coupled to distant functional sites. However, identifying cryptic pockets
remains difficult. Most known cryptic sites were only identified after the serendipitous
discovery of a small molecule that binds and stabilizes the open form of the pocket [241, 74].
Experimental techniques for intentionally identifying and targeting cryptic pockets show great
promise [242, 77, 243], but they still leverage the simultaneous discovery of ligands that bind and
stabilize the open pocket. To overcome this limitation, a number of computational methods have
been developed to identify cryptic pockets without requiring the simultaneous discovery of
small molecules that bind them [47, 49, 244, 245, 246, 247, 248, 249]. These methods
have proved capable of retrodicting a number of previously identified cryptic pockets.
More importantly, applications to a few established drug targets and other enzymes have
successfully identified novel cryptic pockets that have been corroborated by subsequent
experiments [49, 250, 79].
Here, we integrate atomically-detailed computer simulations and biophysical experiments to explore
the potential utility of cryptic pockets in a target that has so far proved undruggable: the interferon
inhibitory domain (IID) of Ebola viral protein 35 (VP35). Ebola virus causes a hemorrhagic fever that
is often lethal, with case fatality rates approaching 90% in past outbreaks [251, 252].
While recent progress in vaccine development and use of biologics, such as antibodies, for
therapeutic and prophylactic purposes show promise [252], small molecule drugs still
offer many advantages, including ease of delivery, lower cost, and longer shelf life. The
1̃20 residue IID of VP35 is a particularly appealing drug target for combating Ebola and
other viruses in the Filoviridae family given that it has a well-conserved sequence and
plays multiple essential roles in the viral lifecycle [253]. One of its primary functions is to
antagonize a host’s innate immunity, particularly RIG-I-like receptor (RLR)-mediated
detection of viral nucleic acids, to prevent an interferon (IFN) response and signaling of
neighboring cells to heighten their antiviral defenses [254, 255, 256]. Crystal structures have
provided a foundation for understanding much about the mechanism of VP35-mediated IFN
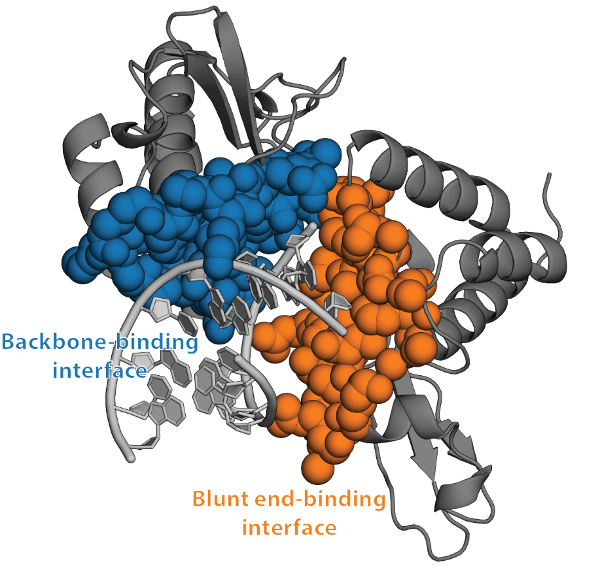
antagonism [257, 258]. For example, they have revealed that VP35’s IID binds both the blunt
ends and backbone of double-stranded RNA (dsRNA), and that there is a PPI between
these dsRNA-binding modes (Fig. 4.1) [258]. Disrupting any of these interactions could
potentially render Ebola susceptible to a host’s innate immunity. In particular, binding
to dsRNA blunt ends plays a dominant role in IFN suppression by Ebola [259]. Indeed,
mutations that reduce the IID’s affinity for dsRNA blunt ends are sufficient to mitigate
IFN antagonism, ultimately attenuating Ebola’s pathogenicity [259, 260, 261, 262]. So,
disrupting this single binding mode could dramatically reduce the impact of an Ebola infection
on the host and potentially reduce deleterious effects, including lethality. However, both
dsRNA-binding interfaces are large flat surfaces that are difficult for small molecules to bind tightly
(Fig. 4.1). As a result, [there are no approved therapies targeting VP35]. Both docking and
screening attempts to discover small molecules that bind these interfaces have not yielded
sufficiently strong leads to warrant clinical development [263, 264]. The discovery of
cryptic pockets in VP35 could provide new opportunities for drugging this essential viral
component.
4.3 Results
4.3.1 Computer simulations reveal a potentially druggable cryptic pocket.
We applied our fluctuation amplification of specific traits (FAST) simulation algorithm [35] to
enhance sampling of structures with large pocket volumes that may harbor cryptic pockets. FAST is a
goal-oriented adaptive sampling algorithm that exploits Markov state model (MSM) methods to focus
computational resources on exploring regions of conformational space with user-specified structural
features. An MSM is a network model of a protein’s energy landscape which consists of a set of
structural states the protein adopts and the rates of hopping between them [265, 53]. Adaptive
sampling algorithms enable efficient construction of MSMs by iteratively 1) running a batch of
simulations, 2) building an MSM, and 3) selecting a subset of the states that have been
identified so far as starting points for the next batch of simulations to maximize the chances
of improving the model [266, 267]. FAST selects which states to further simulate in a
manner that balances exploration/exploitation tradeoffs by considering 1) how well each state
optimizes a user defined structural criterion (in this case maximizing the total pocket volume)
and 2) the likelihood of discovering new conformational states [35]. After running FAST,
we collected additional simulation data by launching each state on the Folding@home
distributed computing environment, which brings together the computing resources of tens of
thousands of citizen scientists who volunteer to run simulations on their personal computers.
Our final model has 11,891 conformational states, providing a detailed characterization
of the different structures the IID adopts but making manual interpretation of the model
difficult.
To identify cryptic pockets within the large ensemble captured by our MSM, we applied our exposons
analysis pipeline [49]. An exposon is a cluster of residues that undergo cooperative changes in their
solvent exposure. Coupling between the solvent exposure of every pair of residues is quantified using
a mutual information metric, as described in Methods. Exposons are often associated with cryptic sites
because the opening/closing of such pockets gives rise to cooperative increases/decreases in the
solvent exposure of surrounding residues. Importantly, once an exposon has been identified, our MSM
framework provides a facile means to identify the conformational changes that give rise to that
exposon.
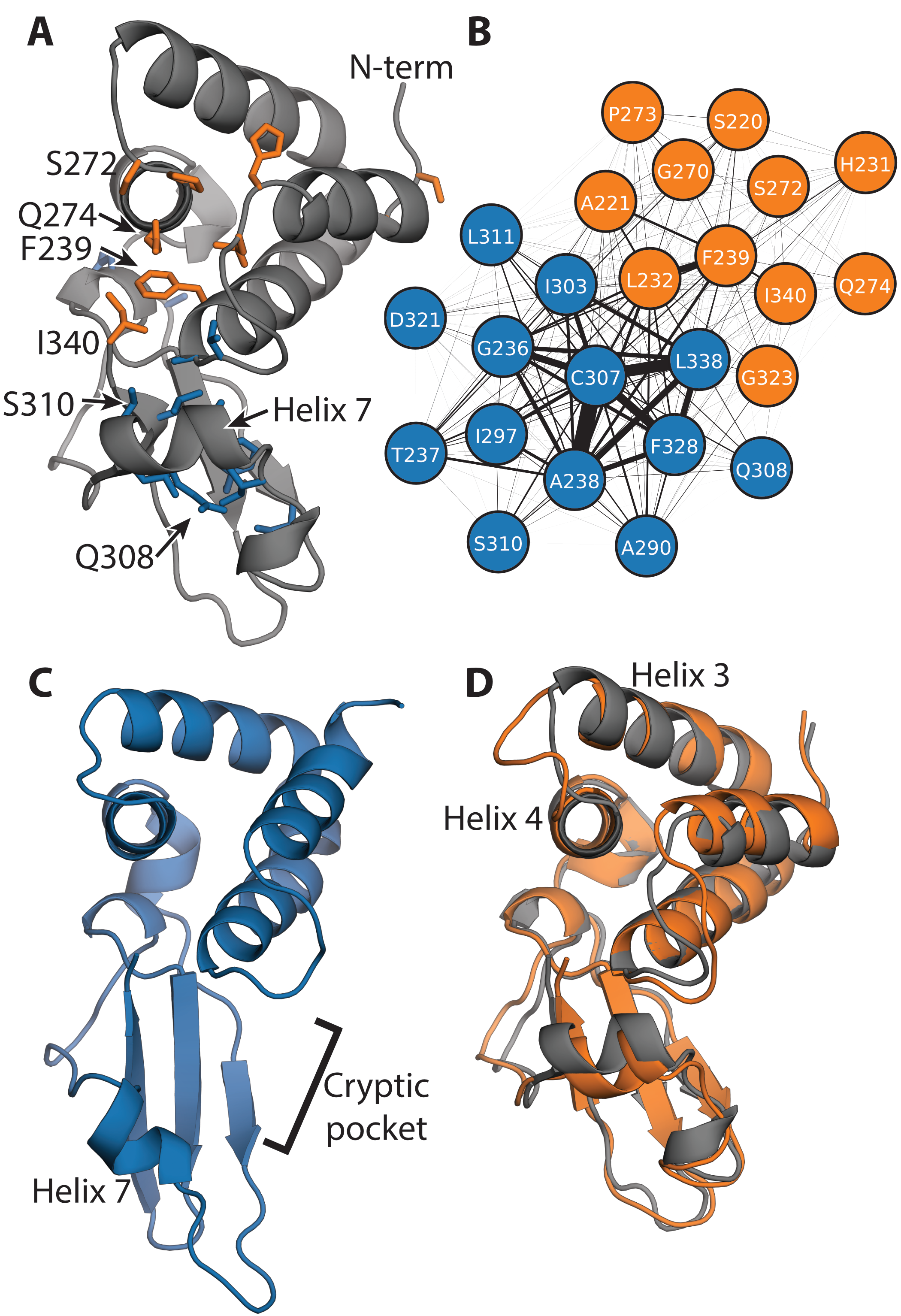
The IID has two significant exposons, one of which corresponds to a large cryptic pocket. The blue
exposon (Fig. 4.2A and 4.2B) consists of a set of strongly-coupled residues in helix 7 and adjacent
loops and secondary structure elements. Visualizing the conformational change that gives rise to
this exposon reveals a substantial displacement of helix 7, creating a large cryptic pocket
between it and the helical domain (Fig. 4.2C). A number of residues that are displaced along
with helix 7 (i.e. A306, K309, and S310) make van der Waals contacts with the dsRNA
backbone in the dsRNA-bound crystal structure [258], so targeting this cryptic pocket could
directly disrupt this binding mode. Retrospective analysis of other validated drug targets
suggests cryptic sites created by the movement of secondary structure elements, such as the
displacement of helix 7, are often druggable [268]. The potential druggability of this cryptic
site is also supported by application of the FTMap algorithm [269, 270], which predicts
a number of hotspots within the pocket where small molecules could form a variety of
energetically-favorable interactions (C.1). Unfortunately, disrupting backbone binding is of less
therapeutic utility than disrupting blunt end binding and it is unknown whether the contacts
between A306, K309, and S310 are essential for backbone binding. Therefore, it is unclear
from this analysis alone whether drugging this newly discovered cryptic pocket would be
useful.
The second exposon (orange in Fig. 4.2) encompasses portions of both dsRNA-binding interfaces, but
it does not correspond to a cryptic pocket. This exposon includes residues that bind dsRNA’s
backbone (i.e. S272) and residues that interact with both the blunt ends and backbone of dsRNA (i.e.
F239, Q274, and I340) [258]. Therefore, altering the conformational preferences of the second
exposon could potentially disrupt the blunt end-binding mode and its crucial role in Ebola’s
ability to evade an immune response. However, the largest conformational change involved
in the formation of this exposon is a displacement of the loop between helices 3 and 4
(Fig. 4.2D). This rearrangement does not create a cryptic pocket that is large enough to
accommodate drug-like molecules, so it is not obvious how to directly manipulate the orange
exposon.
4.3.2 The cryptic pocket is allosterically coupled to the blunt end-binding interface.
Even though the cryptic pocket does not coincide with the interface of VP35’s IID that binds dsRNA
blunt ends, it could still serve as a cryptic allosteric site that allosterically controls RNA
binding. Indeed, the physical proximity of the two exposons and the coupling between them
both hint at the possibility for allosteric coupling. Furthermore, our exposons analysis
could easily underestimate this coupling given that it focuses on correlated transitions of
residues between solvent exposed and completely buried states, leaving it blind to more subtle
conformational fluctuations and the coupling of residues that are always buried (or always
exposed).
To explore the potential for a broader allosteric network, we applied the correlation of all rotameric
and dynamical states (CARDS) algorithm [90]. CARDS classifies each dihedral in each
snapshot of a simulation as being in one of three rotameric states (gauche+, gauche-, or
trans) and one of two dynamical states (ordered or disordered). A dihedral is said to be
disordered if it is rapidly hopping between different structural states, and it is classified as
ordered if it appears to be locked into a single rotameric state for a prolonged time. The
mutual information metric is then used to quantify how strongly coupled the structural and
dynamical states of each pair of dihedrals are, enabling CARDS to capture the roles of both
concerted structural changes and conformational entropy in allosteric communication.
Importantly, CARDS accounts for the potential role of residues that are always buried or always
exposed to solvent and subtle conformational changes that do not alter the solvent exposure of
residues.
CARDS reveals a broader allosteric network than that identified by our exposons analysis and
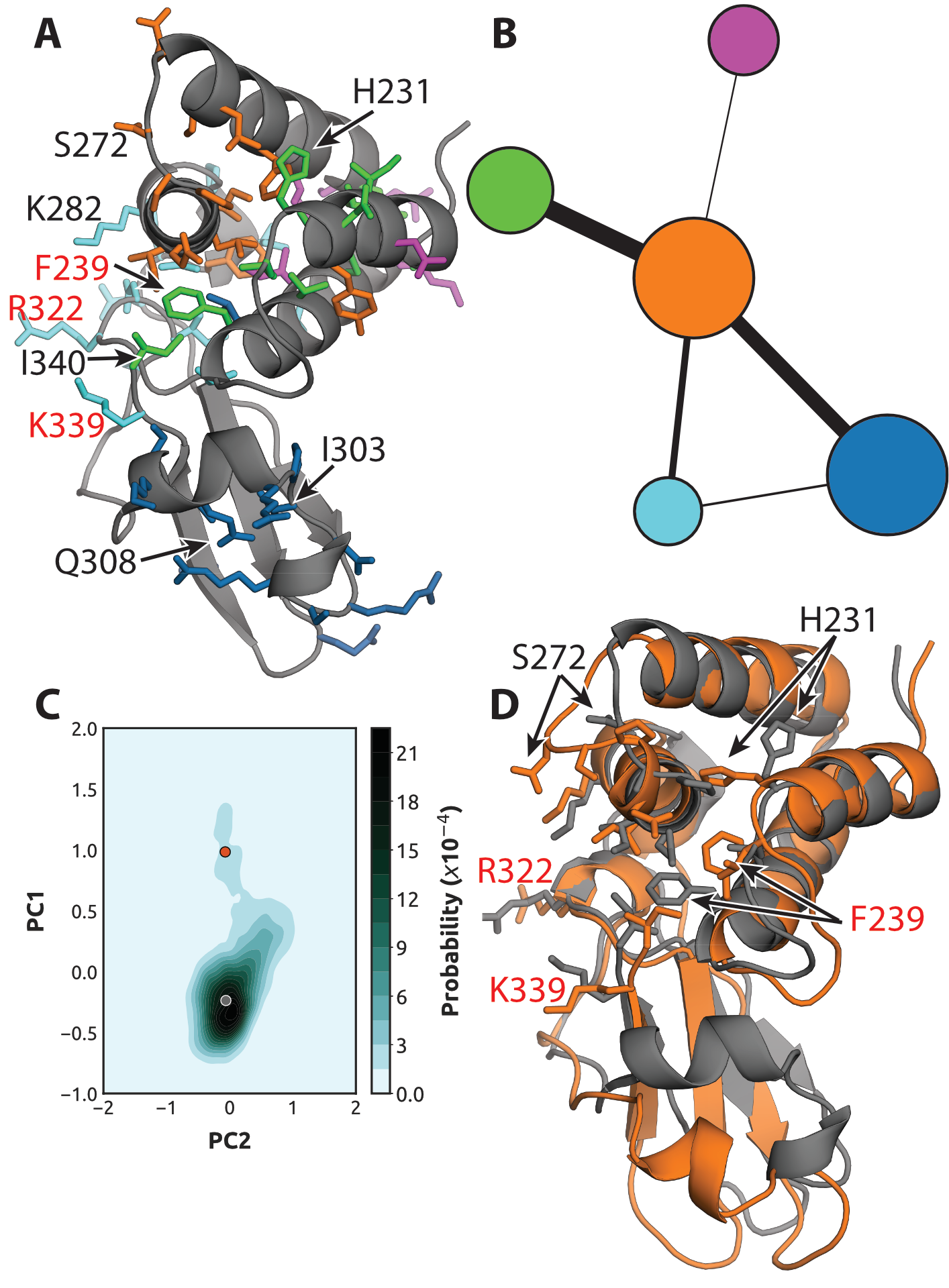
suggests strong coupling between the cryptic pocket and blunt end-binding interface (Fig. 4.3). This
network consists of five communities of strongly coupled residues, four of which coincide with large
portions of the two dsRNA-binding interfaces. One of these communities (orange) is a hub in the
network, having significant coupling to all the other communities. It encompasses part of the
orange exposon, particularly residues around the loop between helices 3 and 4. The orange
CARDS community and exposon both capture Q274, which engages in both dsRNA-binding
interfaces, and S272, which contacts the backbone [258]. However, the CARDS community
includes many additional residues not captured by exposons analysis. Examples include
I278, which engages in both dsRNA-binding interfaces, and D271, which is part of the PPI
between the two binding modes [258]. One of the orange community’s strongest allosteric
connections is to the green community. This community encompasses the rest of the residues
in the orange exposon, including F239 and I340, which are part of both dsRNA-binding
interfaces [258]. The green community also captures additional residues, reaching deep into the
helical domain. The orange community is also strongly coupled to the blue community,
which includes much of helix 7 and nearby residues that move to give rise to the cryptic
pocket that was captured by the blue exposon. Notably, the orange and blue communities are
both coupled to a cyan cluster that was not hinted at by our exposons analysis because the
residues involved are always solvent exposed. It includes R322, which is part of the blunt
end-binding interface and the PPI between the two binding modes, and K282, which also
contacts dsRNA blunt ends [258]. In addition, this community includes K339, which is an
important determinant of the electrostatic favorability of dsRNA binding [258]. Together,
these results suggest that opening of the cryptic pocket could strongly impact residues
involved in both dsRNA-binding interfaces, as well as the PPI between the two binding
modes.
To determine the relative importance of the structural and dynamical preferences of this
community, we compared the magnitudes of the structural and dynamical components of
CARDS. This analysis revealed that concerted structural changes are the dominant mode of
allosteric communication in the IID, rather than conformational entropy and dynamical
allostery (C.2). Therefore, examining structures where the orange community undergoes large
conformational changes might reveal the perturbations these motions induce elsewhere in the
protein.
To understand the potential impact of targeting the cryptic pocket on the blunt end-binding mode, we
performed a dimensionality reduction based on the orange community. Since the orange
community is a hub in the allosteric network, we reasoned that performing a dimensionality
reduction based on the structural preferences of this community and examining representative
structures would report on what is happening throughout the protein. To understand what sort
of conformational changes are present, we performed a dimensionality reduction on our
simulation dataset by applying principal component analysis (PCA) to the distances between the
C
atoms of every pair of residues in the orange community. Projecting our MSM onto the first two
principal components (PC1 and PC2) reveals one dominant free energy minimum and a broad excited
state (Fig. 4.3C).
Comparing representative structures for the orange community’s two dominant states suggests the
cryptic pocket is indeed a cryptic allosteric site, targeting of which could allosterically disrupt binding
of VP35’s IID to dsRNA blunt ends. Most importantly, conformational changes of the orange
community are associated with opening of the cryptic pocket (Fig. 4.3D). Therefore, targeting
the cryptic pocket could modulate the entire allosteric network in addition to its potential
direct effect on the backbone-binding mode. Comparing the structures also reveals that
the end of helix 4 frays and the preceding loop, which sits at the PPI between the two
dsRNA-binding modes, is displaced. So, targeting the cryptic pocket could allosterically
modulate this PPI. Finally, we note a substantial reshuffling of residues F239, H231, and
P273 and modest displacements of R322 and K339. Previous work has demonstrated that
F239A, R322A, and K339A substitutions are each sufficient to disrupt dsRNA binding
and IFN suppression [258]. CARDS analysis suggests targeting the cryptic pocket could
allosterically alter the structures of these residues and have a similar impact on dsRNA
binding.
4.3.3 Thiol labeling experiments corroborate the predicted cryptic pocket.
One way to experimentally test our prediction of a cryptic pocket is to probe for solvent exposure of
residues that are buried in available crystal structures but become exposed to solvent upon pocket
opening. Cysteines are particularly appealing candidates for such experiments because 1) they have a
low abundance and 2) their thiol groups are highly reactive, so it is straightforward to detect
exposed cysteines by introducing labeling reagents that covalently bind accessible thiols.
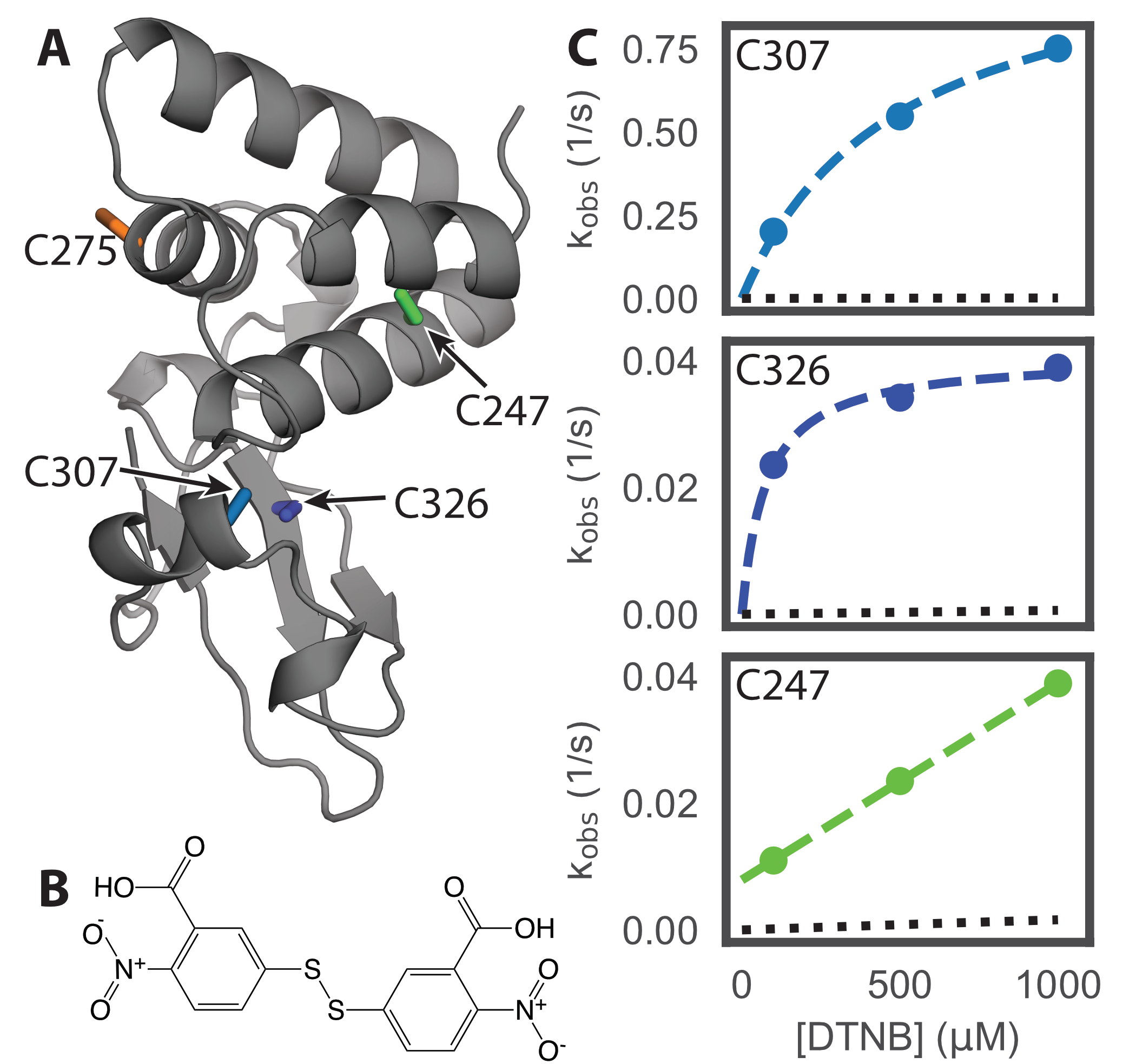
Fortuitously, VP35’s IID has two cysteines (C307 and C326) that are buried in available
crystal structures but become exposed to solvent when the cryptic pocket opens (Fig. 4.4A).
There is also a cysteine (C275) that is on the surface of the apo crystal structure [257]
and a fourth cysteine (C247) that is buried in the helical bundle. C275 is typically solvent
exposed in our simulations, as expected based on the crystallographic data. Examining
the solvent exposure of C247 revealed it is sometimes exposed to solvent via an opening
of helix 1 relative to the rest of the helical bundle (C.3), but FTMap did not identify any
hotspots that are likely to bind drug-like molecules in this region. Therefore, we expect to
observe labeling of all four cysteines on a timescale that is faster than global unfolding of the
protein.
To experimentally test our predicted pocket, we applied a thiol labeling technique that probes the
solvent exposure of cysteine residues [271]. For these experiments, 5,5’-Dithiobis-(2-Nitrobenzoic
Acid) (also known as DTNB or Ellman’s reagent, Fig. 4.4B) is added to a protein sample.
Upon reaction with the thiol group of an exposed cysteine, DTNB breaks into two TNB
molecules, one of which remains covalently bound to the cysteine while the other is released
into solution. The accumulation of free TNB can be quantified based on the increased
absorbance at 412 nm. We have previously applied this technique to test predicted pockets in
-lactamase
enzymes [49, 147].
As expected from our computational model, the observed signal from our thiol labeling experiments is
consistent with opening of the cryptic pocket (Fig. 4.4C). Absorbance curves are best fit by four
exponentials, each with an approximately equivalent amplitude that is consistent with expectations
based on the extinction coefficient for DTNB (C.4). To assign these labeling rates to individual
cysteines, we systematically mutated the cysteines to serines, performed thiol labeling experiments,
and assessed which rates disappeared and which remained (C.6). For example, labeling of the C275S
variant lacks the very fastest rate for wild-type, consistent with the intuition that a residue that is
surfaced exposed in the crystal structure (i.e. C275) should label faster than residues that are generally
buried. To test whether the observed labeling could be due to an alternative process, such as
global unfolding, we determined the population of the unfolded state and unfolding rate of
VP35’s IID under native conditions (C.7) and the intrinsic labeling rate for each cysteine
(C.6). As shown in Fig. 4.4C, the observed labeling rates are all considerably faster than the
expected labeling rate from the unfolded state at a range of DTNB concentrations. This result
confirms that labeling of all four cysteines arises from fluctuations within the native state,
consistent with our computational predictions. Furthermore, the exposure of C247 is far
rarer than C307 or C326 (equilibrium constants for the exposure of C247 and C307 are
5.4×108.110
and 8.5102.810,
respectively). Therefore, a ligand would have to pay a greater energetic cost to stabilize the
conformational change that exposes C247 than to stabilize the open state of the cryptic allosteric site
created by the motion of helix 7.
4.3.4 Stabilizing the open cryptic pocket allosterically disrupts binding to dsRNA blunt
ends.
We reasoned that covalent attachment of TNB to C307 and C326 would provide a means to capture
the open pocket and assess the impact of stabilizing this state on dsRNA binding. Addition of TNB to
these cysteines is sterically incompatible with the closed conformation of VP35’s IID that has been
observed crystallographically. TNB’s mass of 1̃98 Da is also similar to many drug fragments used in
screening campaigns, making it a reasonable surrogate for the type of effect one might achieve with a
fragment hit. Given that we already know DTNB labels the IID’s cysteines, a TNB-labeled sample is
easily obtainable by waiting until the labeling reaction goes to completion. Finally, we have previously
used this same strategy to identify cryptic pockets that exert allosteric control over the activity of
-lactamase
enzymes [49, 147]. To ensure that we primarily capture the effect of labeling on pocket
opening, we used a C247S/C275S variant of VP35’s IID that only has cysteines pointing
into the cryptic pocket. As with the wild-type protein, thiol labeling of the C247S/C275S
variant is consistent with the formation of the proposed cryptic pocket (Supplementary Fig.
C.5).
To measure the effect of TNB labeling on the IID’s interaction with dsRNA, we developed a
fluorescence polarization (FP) assay for monitoring dsRNA binding. Paralleling our past work on
VP35-peptide interactions [272], we added varying concentrations of C247S/C275S IID to a fixed
concentration of 25-bp dsRNA with a fluorescein isothiocyanate (FITC) conjugation at one end (C.7).
Free FITC-dsRNA emits depolarized light upon excitation with polarized light because of the
molecule’s fast rotation. Binding of one or more VP35 molecules restricts the motion of FITC-dsRNA,
resulting in greater emission of polarized light, which is best monitored by the change in
anisotropy [273].
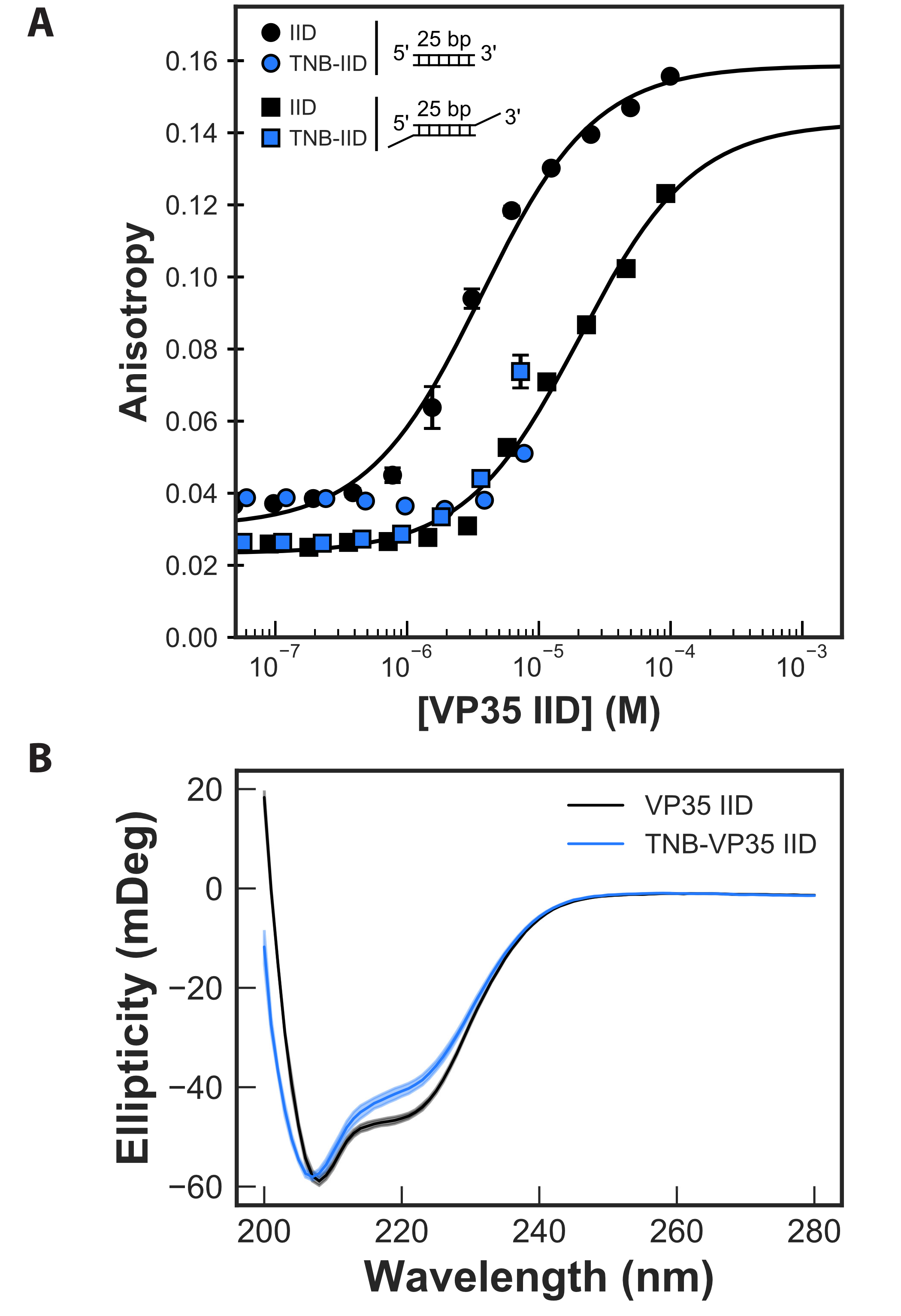
Monitoring the binding of unlabeled protein to 25-bp dsRNA with either blunt ends
or 3’ overhangs demonstrates that our FP assay is sensitive to both dsRNA-binding
modes and gives affinities that are consistent with past work. Past work using a
dot-blot assay to measure binding reported an apparent dissociation constant
(K) for blunt-ended
dsRNA of 3.40.07
M [259].
Furthermore, sterically hindering binding of the IID to dsRNA blunt ends by adding 2-nucleotide
overhangs to the 3’ of the RNA reduces the apparent dsRNA-binding affinity by 10-fold [274]. This
weaker interaction was attributed to the backbone-binding mode since it is still available to VP35’s IID
even when the presence of an overhang inhibits blunt end binding. Similarly, our FP assay gives an
apparent K
of 3.60.34
M for
blunt-ended dsRNA (Fig. 5A). Addition of 3’ overhangs results in a strong rightwards shift of the
binding curve, consistent with at least a 5-fold reduction in the apparent binding affinity (apparent
K of
20.41.1
M).
However, an upper baseline could not be captured due to limitations in the protein’s solubility, so
this apparent Kd is a lower bound. The data are also fit well assuming an apparent Kd of
30.17.2
M that
was reported previously [274].
Repeating our FP assay with TNB-labeled protein reveals that labeling allosterically reduces the
affinity for blunt-ended dsRNA by at least 5-fold (Fig. 4.5A). Solubility limitations again prevented us
from observing complete binding curves for labeled protein, but the data are sufficient to demonstrate
that TNB-labeling has at least as strong an effect on binding as addition of a 3’ overhang. As a control
to ensure that labeling does not disrupt binding by simply unfolding the protein, we measured the
circular dichroism (CD) spectra of labeled and unlabeled protein. The similarity between the CD
spectra (Fig. 4.5B) demonstrates that the IID’s overall fold is not grossly perturbed. The slight
decrease in helicity at 220 nm can be attributed to the covalent modification disrupting the
stability of helix 5 potentially causing local unfolding of this motif. Since the cryptic pocket
does not coincide with the blunt end-binding interface, our results suggests the impact
on dsRNA binding is allosteric. Furthermore, past work demonstrated that reducing the
blunt end-binding affinity by as little as 3-fold is sufficient to allow a host to mount an
effective immune response [258], so targeting our cryptic pocket could be of great therapeutic
value.
4.4 Discussion.
We have identified a cryptic allosteric site in the IID of the Ebola VP35 protein that provides a new
opportunity to target this essential viral component. Past work identified several sites within the VP35
IID that are critical for immune evasion and viral replication [253, 256, 261, 262], but structural
snapshots captured crystallographically lacked druggable pockets [257, 258]. We used adaptive
sampling simulations to access more of the ensemble of conformations that VP35 adopts, uncovering
an unanticipated cryptic pocket. While the pocket directly coincides with the interface that binds the
backbone of dsRNA, it was not clearly of therapeutic relevance since binding dsRNA’s blunt ends is
more important for Ebola’s immune evasion mechanism [259]. However, our simulations also
suggested the cryptic pocket is allosterically coupled to the blunt end-binding interface and,
therefore, could modulate this biologically-important interaction. Subsequent experiments
confirmed that fluctuations within the folded state of the IID expose two buried cysteines
that line the proposed cryptic pocket to solvent. Moreover, covalently modifying these
cysteines to stabilize the open form of the cryptic pocket allosterically disrupts binding to
dsRNA blunt ends by at least 5-fold. Previous work demonstrated that reducing the binding
affinity by as little as 3-fold is sufficient to allow a host to mount an effective immune
response [258]. Therefore, it may be possible to attenuate the impact of viral replication and restrict
pathogenicity by designing small molecules to target the cryptic allosteric site we report
here.
More generally, our results speak to the power of simulations to provide simultaneous access to both
hidden conformations and dynamics with atomic resolution. Such information is extremely difficult to
obtain from single structural snapshots or powerful techniques that report on dynamics without
directly yielding structures, such as NMR and hydrogen deuterium exchange. As a result, simulations
are a powerful means to uncover unanticipated features of proteins’ conformational ensembles, such
as cryptic pockets and allostery, providing a foundation for the design of further experiments. We
anticipate such simulations will enable the discovery of cryptic pockets and cryptic allosteric sites in
other proteins, particularly those that are currently considered undruggable. Furthermore, the detailed
structural insight from simulations will facilitate the design of small molecule drugs that target these
sites.
4.5 Methods
4.5.1 Molecular dynamics simulations and analysis
Simulations were initiated from chain B of PDB 3L25 [258] and run with Gromacs [145] using the
amber03 force field [146] and TIP3P explicit solvent [143] at a temperature of 300 K and 1 bar
pressure, as described previously [9]. We first applied our FAST-pockets algorithm [35] to balance 1)
preferentially simulating structures with large pocket volumes that may harbor cryptic
pockets with 2) broad exploration of conformational space. For FAST, we performed 10
rounds of simulations with 10 simulations/round and 80 ns/simulation. To acquire better
statistics across the landscape, we performed an RMSD-based clustering using a hybrid
k-centers/k-medoids algorithm [230] implemented in Enspara [39] to divide the data into
1,000 clusters. Then we ran three simulations initiated from each cluster center on the
Folding@home distributed computing environment, resulting in an aggregate simulation time of 122
s.
Exposons were identified using our previously described protocols,11 as implemented in
Enspara [39]. Briefly, the solvent accessible surface area (SASA) of each residue’s side-chain was
calculated using the Shrake-Rupley algorithm [275] implemented in MDTraj [148] using a
drug-sized probe (2.8 Å sphere). Conformations were clustered based on the SASA of each residue
using a hybrid k-centers/k-medoids algorithm, using a 2.5 Å2 distance cutoff and 5 rounds of
k-medoids updates. A Markov time of 6 ns was selected based on the implied timescales
test (C.8). The center of each cluster was taken as an exemplar of that conformational
state, and residues were classified as exposed if their SASA exceeded 2.0 Å2 and buried
otherwise. The mutual information between the burial/exposure of each pair of residues
was then calculated based on the MSM (i.e. treating the centers as samples and weighting
them by the equilibrium probability of the state they represent). Finally, exposons were
identified by clustering the matrix of pairwise mutual information values using affinity
propagation [276].
The CARDS algorithm [90] was applied to identify allosteric coupling using our established
protocols [48], as implemented in Enspara [90]. Briefly, each dihedral angle in each snapshot of the
simulations was assigned to one of three rotameric states (gauche+, gauche-, or trans) and one of two
dynamical states (ordered or disordered). The total coupling between each pair of dihedrals
and
was
then calculated as
|
| (4.1) |
where is the mutual
information metric, is the
rotameric state of dihedral ,
and is the dynamical
state of dihedral .
The term
is the purely structural coupling, while the sum of the other three terms is referred to as the
disorder-mediated coupling. The dihedral level couplings were coarse-grained into residue-level
coupling by summing the total coupling between all the relevant dihedrals. Communities of coupled
residues were identified by clustering the residue-level matrix of total couplings using affinity
propagation [276]. The constructed network was subsequently filtered to only retain significant
edges [277].These algorithms are available at github.com/bowman-lab.
4.5.2 Protein expression and purification
All variants of VP35’s IID were purified from the cytoplasm of E. coli BL21(DE3) Gold
cells (Agilent Technologies). Variants were generated using the site directed mutagenesis
method and confirmed by DNA sequencing. Transformed cells were grown at 37℃ until OD
0.3 then grown at 18℃ until induction at OD 0.6 with 1 mM IPTG (Gold Biotechnology,
Olivette, MO). Cells were grown for 15 hours then centrifuged after which the pellet was
resuspended in 20 mM Sodium Phosphate pH 8, 1 M sodium chloride, with 5.1 mM
-mercaptoethanol.
Resuspended cells were subjected to sonication at 4<℃ followed by centrifugation. The supernatant
was then subjected to Ni-NTA affinity, TEV digestion, cation exchange (BioRad UNOsphere Rapid S
column), and size exclusion chromatography (BioRad Enrich SEC 70 column) into 10 mM Hepes pH
7, 150 mM NaCl, 1 mM MgCl2, 2 mM TCEP.
4.5.3 Thiol labeling
We monitored the change in absorbance over time of 5,5’-dithiobis-(2-nitrobenzoic acid) (DTNB,
Ellman’s reagent, Thermo Fisher Scientific). Various concentrations of DTNB were added to protein
and change in absorbance was measured in either an SX-20 Stopped Flow instrument (Applied
Photophysics, Leatherhead, UK), or an Agilent Cary60 UV-vis spectrophotometer at 412 nm until the
reaction reached steady state (3̃00 s). Data were fit with a Linderstrøm-Lang model to extract the
thermodynamics and/or kinetics of pocket opening, as described in detail previously [49]. As a
control, the equilibrium constant for folding and the unfolding rate were measured (C.1)
and used to predict the expected labeling rate from the unfolded state. The equilibrium
constant was inferred from a two-state fit to urea melts monitored by fluorescence and
unfolding rates were inferred from single exponential fits to unfolding curves monitored by
fluorescence after the addition of urea, as described previously [49, 147, 278]. Fluorescence
data were collected using a Photon Technology International Quanta- Master 800 rapid
excitation spectrofluorometer with Quantum Northwest Inc. TC-125 Peltier-controlled cuvette
holder.
4.5.4 Fluorescence polarization binding assay
Binding affinities between variants of VP35’s IID and dsRNA were measured using fluorescence
polarization in 10 mM Hepes pH 7, 150 mM NaCl, 1 mM MgCl2. A 25 base pair FITC-dsRNA
(Integrated DNA Technologies) substrate with and without a 2 nucleotide 3’ overhang was included at
100 nM. The sample was equilibrated for one hour before data collection. Data were collected on a
BioTek Synergy2 Multi-Mode Reader as polarization and were converted to anisotropy as described
previously [273]. TNB-labeled samples were generated by allowing DTNB and VP35’s
IID to react for 3 minutes and then removing excess DTNB with a Zeba spin desalting
columns (Thermo Fisher Scientific). A single-site binding model was sufficient to fit the
data.
4.6 Acknowledgements
We are grateful to the citizen scientists who participate in Folding@home for volunteering to
run simulations on their personal computers. This work was funded by NSF CAREER
Award MCB-1552471 and NIH grant R01 GM124007 (Bowman), as well as NIH grants
R01AI123926, P01AI120943, and R01AI143292 (Amarasinghe). GRB holds a Career Award at
the Scientific Interface from the Burroughs Wellcome Fund and a Packard Fellowship
for Science and Engineering from The David & Lucile Packard Foundation. MAC was
supported by the 5R25GM103757 IMSD program and SS was supported by a MilliporeSigma
Fellowship. We thank Drs. Timothy M. Lohman and Alexander G. Kozlov for advice on FP
assays.